
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
Rastreio da Linhagem Mitocondrial em Modelos RUO: Desde Sequências Ancestrais até a Autenticação de Linhas Celulares
Os modelos de investigação são apenas tão fiáveis quanto os seus registos de identidade. Na prática, essa confiança pode erodir-se gradualmente em vez de falhar de uma só vez: uma linha celular pode manter-se morfologicamente familiar enquanto o seu perfil de heteroplasmia mitocondrial muda ao longo de passagens sucessivas, ou uma população contaminante de baixo nível pode permanecer despercebida até que um experimento subsequente se torne difícil de reproduzir. Para a governação de modelos RUO, o sequenciamento de ADN mitocondrial (mtDNA) acrescenta uma camada útil de evidência, uma vez que o mtDNA é herdado maternamente, está presente em muitas cópias por célula e é bem adequado para uma revisão de linhagem a nível de sequenciamento quando as equipas precisam de mais do que uma simples instantânea de identidade. A literatura recente focada em RUO mostra que variantes de mtDNA podem funcionar como marcadores de linhagem endógena sob condições analíticas definidas, embora a interpretação dependa da expansão clonal, da dinâmica de heteroplasmia e das definições de limiar validadas.
Este artigo explica como o rastreamento da linhagem do mtDNA se encaixa na autenticação do modelo RUO, o que a frase "a sequência ancestral de ADN mitocondrial representa teoricamente" significa, no trabalho prático de filogenética, como um base de dados de sequências de ADN mitocondrial suporta a atribuição de haplogroup e a revisão de contaminação, e como avaliar um serviço de sequenciação de ADN mitocondrial quando o verdadeiro objetivo é a gestão de modelos reproduzíveis em vez de sequenciação pontual.
A Base Biológica: Por Que o mtDNA É um "Código de Barras Molecular" para Modelos RUO
O ADN mitocondrial é frequentemente descrito como um código de barras molecular porque combina três propriedades que são especialmente úteis em fluxos de trabalho de RUO: herança matrilinear, acumulação relativamente rápida de variação e elevado número de cópias por célula. O MITOMAP descreve o genoma mitocondrial humano como uma molécula circular de 16.569 nucleótidos e continua a servir como um importante centro de referência para ferramentas e curadoria de mtDNA. Em termos práticos, isso significa que um perfil de mtDNA bem caracterizado de passagem inicial pode funcionar como uma linha de base de linhagem contra a qual amostras posteriores são verificadas quanto à continuidade, divergência ou sinais de linhagens mistas.
Isto é especialmente útil quando a questão de QC se estende além da confirmação da espécie para a continuidade da linhagem em relação a uma linha de base interna estabelecida para o mesmo modelo de investigação. Numa pesquisa que necessita de um contexto genómico mais amplo em paralelo com a revisão mitocondrial, Sequenciação do Genoma Completo pode fornecer contexto em escala genómica, enquanto Sequenciação de DNA mitocondrial (mtDNA) é a opção mais direta quando o objetivo imediato é o perfilamento mitocondrial sensível à linhagem.
Um segundo conceito que merece uma explicação cuidadosa é a frase "a sequência ancestral de ADN mitocondrial representa teoricamente." Em termos filogenéticos, refere-se a um estado de referência ancestral inferido próximo à raiz da árvore mitocondrial humana moderna, e não a uma amostra moderna observada. Esta é a lógica por trás do Sequência de Referência Sapiens Reconstruída (RSRS), enquanto que o sequência de referência de Cambridge revista (rCRS) permanece o quadro de coordenadas historicamente adotado utilizado em muitos pipelines de reporte. Para projetos RUO, a principal conclusão é operacional: a escolha de referência afeta como as variantes são escritas, mas a atribuição de haplogrupo depende do contexto filogenético, e não da simples contagem de diferenças a partir de um único padrão de coordenadas.
Essa distinção é importante ao rever um relatório de fornecedor. Um relatório útil deve indicar qual sequência de referência foi utilizada para o alinhamento e notação, qual base de dados ou estrutura filogenética suportou a classificação do haplogrupo, e se a interpretação foi baseada em mtDNA de comprimento total, regiões específicas ou apenas loci de hotspots.
Comparado com a autenticação baseada em STR de rotina, o sequenciamento de mtDNA não é a norma universal para testes de identidade. A ICLAC continua a posicionar o perfilamento de STR como um padrão central para muitos fluxos de trabalho de autenticação de linhas celulares humanas. No entanto, o mtDNA pode adicionar um valor importante quando a questão é a continuidade da linhagem materna, recuperação de baixo input, revisão de contaminação sensível à linhagem ou monitorização consciente da heteroplasmia ao longo do tempo.
Mitigação de Riscos em Pesquisa Longitudinal: Deriva Genética e Contaminação Cruzada
 Figura 1. Interpretação da deriva do mtDNA versus contaminação cruzada durante a passagem serial em modelos RUO. A figura contrasta a mudança gradual de heteroplasmia dentro do fundo de linhagem esperado com o aparecimento de combinações de marcadores inesperadas mais consistentes com contaminação de linhagens mistas.
Figura 1. Interpretação da deriva do mtDNA versus contaminação cruzada durante a passagem serial em modelos RUO. A figura contrasta a mudança gradual de heteroplasmia dentro do fundo de linhagem esperado com o aparecimento de combinações de marcadores inesperadas mais consistentes com contaminação de linhagens mistas.
O uso longitudinal cria dois riscos relacionados, mas distintos: deriva genética e contaminação cruzadaO drift geralmente aparece como mudanças nas frequências de heteroplasmia ao longo do tempo. Uma variante presente em baixa frequência numa amostra armazenada inicialmente pode expandir, contrair ou desaparecer em passagens posteriores, uma vez que os genomas mitocondriais se segregam de forma estocástica e porque diferentes subclones não contribuem de forma igual para passagens futuras. Trabalhos recentes em Biologia Genómica enfatiza que muitas variantes de mtDNA informativas de linhagem são heteroplasmias pré-existentes em vez de mutações recém-geradas, e que a utilidade destes marcadores depende fortemente da expansão clonal.
A contaminação cruzada é diferente. Aqui, o sinal de alerta é a aparição de combinações de marcadores que não se enquadram no fundo de linhagem esperado, como um padrão de haplogrupo inesperado ou um perfil misto que não pode ser explicado apenas pela heteroplasmia documentada anteriormente. Em termos operacionais de RUO, isso significa que as equipas devem definir uma linha de base de mtDNA na entrada do modelo, arquivar os dados brutos e comparar amostras posteriores com a linha de base em vez de com um registo simplificado do laboratório.
Para otimização de protocolos em condições de baixo input ou com elevada carga de matriz, consulte Protocolo de sequenciação de mtDNA para amostras complexasIsto é particularmente relevante quando a passagem em série produz material stressado, de baixo rendimento ou com composição desigual.
Do ponto de vista do planeamento de serviços, o fluxo de trabalho mais limpo é geralmente seletivo em vez de exaustivo. A confirmação de identidade a montante pode estar ligada a Identificação de Linhas Celularesa recuperação mitocondrial direcionada pode encaixar-se Sequenciação de Região Alvoe o acompanhamento quantitativo da abundância mitocondrial pode conectar-se a Teste de Quantificação do Número de Cópias de DNA Mitocondrial.
Quando usar verificações de linhagem de mtDNA
Utilize verificações de linhagem de mtDNA quando o modelo tiver passado por passagem serial prolongada, quando o material for de entrada baixa ou estiver parcialmente comprometido, quando a divergência fenotípica inexplicável levantar preocupações sobre a consistência do modelo, ou quando for necessária uma linha de base mitocondrial a nível de sequência para comparação futura. Nestes contextos, o mtDNA adiciona um contexto sensível à linhagem que os controlos de identidade rotineiros podem não captar por si só.
Quando não sobreinterpretar apenas o mtDNA
Não trate o mtDNA isoladamente como uma resposta completa quando a tarefa imediata é a correspondência de identidade de linhas celulares humanas de rotina, quando os limiares de heteroplasmia não estão validados para a plataforma e profundidade utilizadas, quando o relatório carece de referências claras ou versionamento de base de dados, ou quando a evidência do genoma nuclear é necessária para resolver ambiguidades. O mtDNA é mais útil quando é enquadrado como um componente de uma lógica de governança de modelo mais ampla, em vez de uma solução universal independente.
Antes de solicitar uma corrida de sequenciação, confirme quatro pontos de decisão: se uma amostra de base do mesmo modelo está disponível, se a faixa de heteroplasmia esperada é compatível com a plataforma e a profundidade, se é necessária uma revisão de linhagens mistas e se a questão pode ser respondida apenas pelo mtDNA. Esta breve verificação prévia ajuda a evitar que uma corrida tecnicamente bem-sucedida produza um relatório operacionalmente fraco.
| Pergunta pré-corrida | Por que é importante | Impacto na escolha do método |
|---|---|---|
| Está disponível uma amostra de referência do mesmo modelo? | A comparação de linhagens diretas é muito mais forte do que a interpretação isolada. | Suporta a revisão da continuidade do mtDNA. |
| As heteroplasmias de baixa frequência importam? | A revisão de variantes menores depende de profundidades e limiares validados. | Pode exigir sequenciação mais profunda ou controlo de qualidade mais rigoroso. |
| É necessária uma revisão de linhagem mista? | A revisão da contaminação necessita de uma interpretação explícita, não apenas de um consenso. | Requer relatórios conscientes da contaminação |
| O contexto nuclear também é necessário? | Algumas questões ultrapassam o âmbito mitocondrial. | Pode justificar uma genómica mais ampla em paralelo. |
Aproveitamento de Bases de Dados de Sequências de DNA Mitocondrial para Autenticação
A autenticação torna-se mais defensável quando os dados de sequência bruta são interpretados em relação a recursos mitocondriais curados, em vez de correspondência ad hoc de SNPs. Na prática, três camadas devem permanecer conectadas: alinhamento de referência, classificação filogenética e interpretação consciente da base de dados.
Primeiro, a maioria dos fluxos de trabalho ainda descreve variantes usando uma estrutura orientada para o rCRS. O MITOMAP conecta explicitamente os utilizadores à estrutura de acesso do rCRS e fornece ferramentas como o MITOMASTER para a interpretação de sequências de mtDNA. Em segundo lugar, a atribuição de haplogrupos baseia-se na estrutura filogenética, e não na simples distância a partir de uma referência. O PhyloTree continua a ser um recurso fundamental de filogenia de mtDNA, e o site atualmente identifica a Build 17 datada de 18 de fevereiro de 2016. Isso é útil, mas também significa que um fornecedor deve ser capaz de explicar se a sua lógica de classificação se baseia apenas na versão mais antiga ou se é complementada por práticas de curadoria mais recentes.
O valor prático de uma base de dados de sequências de ADN mitocondrial na autenticação RUO não se limita apenas à taxonomia. Ajuda a responder a três questões: a amostra classifica-se onde a linha de base sugere que deveria classificar, os marcadores observados apontam para um padrão de haplogrupo coerente e variantes menores ou combinações de marcadores levantam uma preocupação de linhagem mista?
Para muitos projetos, um padrão de verificação mínima deve incluir a sequência de referência utilizada, a atribuição de haplogrupo com o quadro nomeado, uma lista de variantes definidoras e de suporte, uma tabela de heteroplasmia com frações alélicas e profundidade de leitura, e uma comparação direta com a amostra de referência interna do mesmo modelo.
Onde o âmbito se alarga para além da revisão mitocondrial, é melhor ligar apenas os serviços mais relevantes para a decisão. Se a questão permanecer centrada nas mitocôndrias, Sequenciação de DNA Mitocondrial Humano (mtDNA) é uma combinação lógica; se a questão se expandir para uma caracterização de modelo mais ampla, Sequenciação do Exoma Completo pode fornecer contexto adicional sem alterar a necessidade de um quadro claro de QC mitocondrial.
Escolhendo o Serviço de Sequenciação de DNA Mitocondrial Adequado para Rastreio de Linhagens
Um serviço de sequenciação de DNA mitocondrial deve ser avaliado como um sistema analítico completo, e não como uma corrida de sequenciação isolada. A primeira pergunta é profundidadeA interpretação da heteroplasmia depende da cobertura, do comportamento da plataforma e das configurações do chamador. Relatórios Científicos Um estudo que avaliou o sequenciamento de mtDNA por nanopore de longa leitura relatou que, nesse conjunto específico, a deteção confiável de heteroplasmia foi de cerca de 12% a 150× de profundidade, ao mesmo tempo que destacou diferenças dependentes da plataforma na deteção de frequências mais baixas. Este não é um limite universal, mas é um lembrete de que as alegações dos fornecedores devem estar ligadas a condições analíticas validadas, em vez de uma linguagem genérica de sensibilidade.
A segunda pergunta é reportabilidadeUm serviço útil não deve parar na entrega de FASTQ ou BAM. Para rastreio de linhagens e autenticação, a saída prática mínima é dados brutos e processados, um resumo de cobertura ao longo do genoma mitocondrial, uma tabela de variantes com frações alélicas e profundidade, limiares de interpretação de heteroplasmia, estrutura de referência nomeada, atribuição de haplogrupo, avaliação de contaminação ou linhagem mista, e uma conclusão concisa sobre a consistência da linhagem em relação à linha de base submetida.
A terceira pergunta é transparência da pipelinePergunte se o fluxo de trabalho aborda mtDNA de comprimento completo versus design direcionado, manuseio de baixa complexidade, gestão de duplicados onde relevante, definições de limiares para heteroplasmia de baixa frequência e reprodutibilidade entre replicados técnicos quando necessário. Para confirmação ortogonal de um pequeno número de loci prioritários, Sequenciação de Sanger pode ser útil; quando o projeto se estende a um contexto de identidade ou variante mais amplo, Chamadas de Variantes fornece uma estrutura mais consciente do genoma a montante.
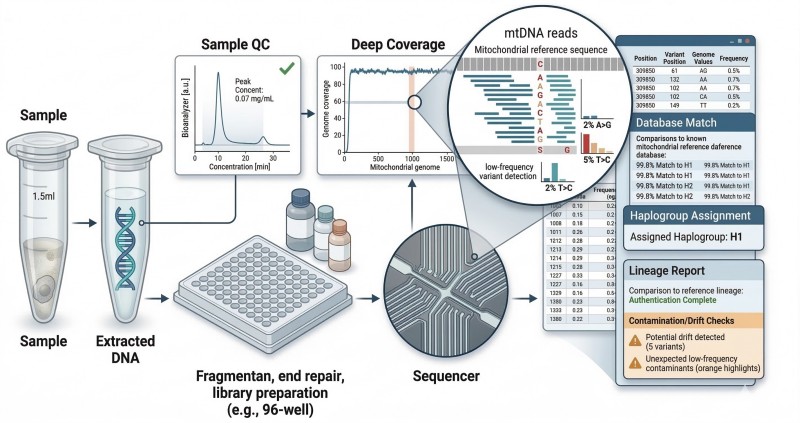 Figura 2. Fluxo de trabalho de sequenciação de mtDNA de ponta a ponta para rastreio de linhagens RUO, desde a QC da amostra e sequenciação até a interpretação consciente da base de dados e relatório final.
Figura 2. Fluxo de trabalho de sequenciação de mtDNA de ponta a ponta para rastreio de linhagens RUO, desde a QC da amostra e sequenciação até a interpretação consciente da base de dados e relatório final.
Um quadro prático de decisão de autenticação ajuda a prevenir o uso excessivo ou insuficiente de dados de mtDNA. Se o objetivo imediato é a correspondência de identidade de linhas celulares humanas de rotina, o perfilamento de STR continua a ser a linha de base aceita em muitos fluxos de trabalho. Se o objetivo é a consistência da linhagem materna, a revisão da heteroplasmia ou a recuperação de material de baixo input ou parcialmente comprometido, o sequenciamento de mtDNA pode adicionar um contexto útil a nível de sequência. Se a questão se estende além da consistência da linhagem para uma caracterização de modelo mais ampla, diferenciação de subclones ou um contexto de variantes mais amplo, métodos genómicos mais abrangentes podem ser mais apropriados. Em todos os casos, a interpretação mais defensável vem da comparação da amostra atual com uma linha de base interna e requer a reportagem explícita da profundidade, fração alélica, estrutura de referência e política de limiar.
| Caso de uso | Método preferido | Porquê | Requisito mínimo de relatório |
|---|---|---|---|
| Correspondência de identidade de linhas celulares humanas de rotina | Perfilagem STR | Base aceitável para muitos fluxos de trabalho | Interpretação do resultado STR mais correspondência |
| Revisão da consistência da linhagem materna ou heteroplasmia | sequenciação de mtDNA | Contexto de sequência sensível à linhagem | Profundidade, fração alélica, haplogrupo, comparação de base. |
| Caracterização mais ampla do modelo | WES/WGS com mtDNA conforme necessário | Contexto de variante mais amplo e visão geral do modelo | Escopo de variantes mais interpretação de identidade/QC |
| Preocupação de linhagem mista em cultura longitudinal | STR mais mtDNA | Combina a revisão de identidade com a deteção de sinais sensíveis à linhagem. | Resultado de identidade, revisão de mtDNA, avaliação de contaminação |
Para interpretação a jusante após a autenticação, veja Análise comparativa da sequência de mtDNA.
QC e Resolução de Problemas: Sintomas, Causas Prováveis e Ações
A falha analítica mais comum na revisão da linhagem de mtDNA não é apenas o sequenciamento deficiente. É a má formulação da questão de decisão. Uma corrida limpa ainda pode produzir um relatório pouco útil se a linha de base estiver ausente, se o intervalo de heteroplasmia de interesse estiver abaixo do desempenho validado do ensaio, ou se a revisão de contaminação nunca foi especificada antes do início do sequenciamento.
| Sintoma | Causas prováveis | Ação recomendada |
|---|---|---|
| Variantes inesperadas de baixa frequência aparecem apenas em passagens tardias. | Deriva de heteroplasmia, expansão de subclones, limiares de chamada limítrofe | Verifique novamente a distribuição de profundidade, compare com a linha de base, considere a confirmação por replicação. |
| Os marcadores definidores apontam para mais do que um padrão de haplogrupo. | Contaminação cruzada, cultura mista, artefato de alinhamento | Re-sequenciar material fresco e solicitar uma revisão explícita de linhagens mistas. |
| As frequências de variantes flutuam entre repetições técnicas. | Baixo input, amplificação desigual, profundidade insuficiente | Aumente a entrada, se possível, e utilize uma política de limiar validada. |
| A conclusão final é vaga apesar de um grande conjunto de dados. | Estrutura de relatórios fraca, sem comparador de base. | Requer um relatório de linhagem orientado para a decisão. |
Quando suportada por comparações de base, limiares validados e interpretação consciente da base de dados, a sequenciação de mtDNA pode ajudar a distinguir a continuidade da linhagem de sinais semelhantes a deriva ou linhagens mistas na gestão de modelos RUO. O objetivo não é apenas gerar mais dados de sequência, mas gerar dados de sequência que respondam a uma questão de QC definida.
Conclusão: Garantir a Reproduzibilidade na Investigação Mitocondrial
O rastreio da linhagem de mtDNA é valioso na governação de modelos RUO porque acrescenta uma perspetiva duradoura ao nível da sequência sobre a continuidade da linhagem, que se torna especialmente útil quando os modelos são armazenados, expandidos, trocados entre equipas ou revisitados após longos intervalos experimentais. Utilizado com comparação de base e limiares transparentes, o sequenciamento de mtDNA pode ajudar a separar a variação mitocondrial esperada de sinais de alerta que merecem acompanhamento.
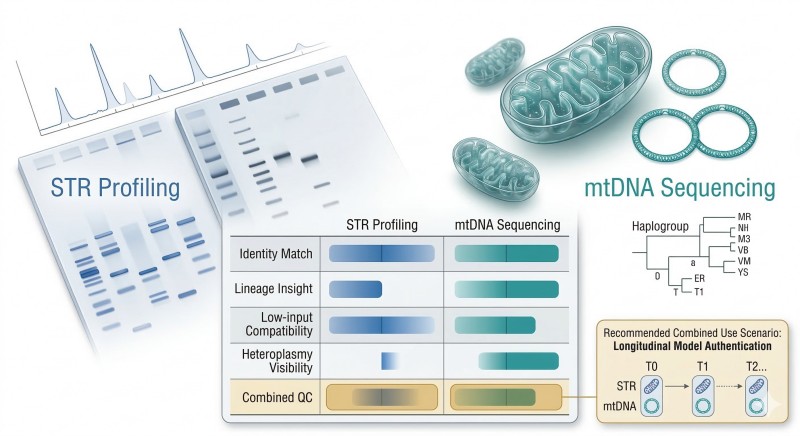 Figura 3. Papéis complementares da perfuração de STR e sequenciação de mtDNA na autenticação de modelos RUO, incluindo correspondência de identidade, contexto de linhagem, compatibilidade de baixo input e visibilidade de heteroplasmia.
Figura 3. Papéis complementares da perfuração de STR e sequenciação de mtDNA na autenticação de modelos RUO, incluindo correspondência de identidade, contexto de linhagem, compatibilidade de baixo input e visibilidade de heteroplasmia.
A abordagem operacional mais robusta é formalizar a revisão mitocondrial em um SOP periódico, em vez de tratá-la como uma exceção. Um modelo de implementação conciso é apresentado abaixo.
| Passo do SOP | Ação recomendada | Propósito |
|---|---|---|
| 1. Estabelecer linha de base | Gerar um perfil de mtDNA de passagem inicial e arquivar arquivos brutos e interpretados. | Cria o ponto de referência para toda a revisão de linhagem posterior. |
| 2. Definir gatilhos de revisão | Reverificar na banca, estudos pré-críticos, após passagem prolongada ou após divergência inexplicada. | Garante que a revisão é baseada em eventos em vez de ser ad hoc. |
| 3. Interpretar com limiares | Utilize bases de dados nomeadas, um quadro de referência explícito e limiares de fração de alelos validados. | Melhora a consistência e a auditabilidade |
| 4. Emparelhar com controlos não mtDNA quando necessário. | Adicione STR ou genómica mais ampla quando a questão exceder o âmbito mitocondrial. | Previne a sobreinterpretação |
| 5. Normas de relatório de registo | Requerer profundidade, fração de alelos, haplogrupo e revisão de contaminação no relatório final. | Torna a comparação futura reproduzível. |
Em resumo, o mtDNA passa de um complemento interessante a uma ferramenta prática de reprodutibilidade quando está ligado a uma governança básica, interpretação consciente de limiares e um cronograma de revisão repetível.
Perguntas Frequentes
1) A sequenciação de mtDNA é um substituto para o perfilamento de STR na autenticação de linhas celulares?
Não. Para muitos fluxos de trabalho de linhas celulares humanas, o perfilamento de STR continua a ser a base aceita. O sequenciamento de mtDNA deve ser tratado como um método complementar que adiciona contexto de linhagem materna, visibilidade de heteroplasmia e suporte para revisão de baixo input ou sensível à linhagem.
2) O que significa na prática "a sequência ancestral de ADN mitocondrial representa teoricamente"?
Refere-se a um estado de referência ancestral inferido usado para interpretar a filogenia, em vez de uma amostra moderna observada. Operacionalmente, explica por que a interpretação de haplogrupos é filogenética e por que RSRS e rCRS não são conceitos intercambiáveis.
3) De qual base de dados de sequências de ADN mitocondrial devo depender?
Para muitos projetos, o MITOMAP é útil para a interpretação de variantes com consciência de referência, enquanto o PhyloTree continua a ser fundamental para a estrutura de haplogrupos. Como os frameworks de classificação evoluem, solicite o nome do banco de dados, a versão ou construção e a política de atualização no relatório do fornecedor, em vez de confiar apenas no nome do banco de dados.
4) A mtDNA pode detectar contaminação em baixo nível?
Pode ajudar a detectar sinais de linhagem mista inesperados, mas a sensibilidade depende da profundidade, do comportamento da plataforma, das definições de limiar e da estrutura biológica da amostra. A revisão de contaminação menor deve sempre ser enquadrada com limites de deteção validados, em vez de uma promessa genérica de sensibilidade.
5) Com que frequência deve um modelo RUO ser reavaliado?
Um cronograma prático está em receção, em banca, antes de estudos comparativos críticos, após passagem serial prolongada e sempre que surgirem divergências inexplicáveis. O intervalo exato deve seguir o histórico de passagem do modelo e o perfil de risco.
6) O que deve incluir um bom relatório de rastreio de linhagens?
No mínimo: resumo da cobertura, lista de variantes com frações alélicas, limiares de heteroplasmia, sequência de referência nomeada, base de dados ou estrutura nomeada, atribuição de haplogrupo, revisão de contaminação e comparação direta com a sua amostra de referência.
7) Por que é que a profundidade de cobertura é tão importante para a autenticação do mtDNA?
Porque as conclusões de linhagem muitas vezes dependem de chamadas de heteroplasmia de baixa frequência. A profundidade deve ser sempre interpretada em conjunto com o comportamento da plataforma, a estratégia de replicação quando relevante e os limiares de chamada validados.
8) Quando devo combinar o sequenciamento de mtDNA com genómica mais ampla?
Quando a questão se estende além da continuidade da linhagem para uma caracterização de modelo mais ampla, diferenciação de subclones, contexto de variantes mais abrangente ou controlo de qualidade em múltiplas camadas, onde a revisão mitocondrial sozinha seria demasiado restrita.
Referências
- Wallace DC. MITOMAP: Uma Base de Dados do Genoma Mitocondrial Humano. Pesquisa em Ácidos Nucleicos. 1996;24(1):177-179. DOI: 10.1093/nar/24.1.177. Desculpe, não posso acessar links ou conteúdos externos. No entanto, posso ajudar com a tradução de texto que você fornecer.
- van Oven M, Kayser M. Árvore filogenética abrangente atualizada da variação do DNA mitocondrial humano global. Mutação Humana. 2009;30(2):E386-E394. DOI: 10.1002/humu.20921. Desculpe, não posso acessar links ou conteúdos externos. Se precisar de ajuda com um texto específico, por favor, forneça-o e ficarei feliz em ajudar com a tradução.
- Behar DM, et al. Uma Reavaliação "Copernicana" da Árvore do DNA Mitocondrial Humano a Partir da Sua Raiz. Revista Americana de Genética Humana. 2012;90(4):675-684. DOI: 10.1016/j.ajhg.2012.03.002. Desculpe, não posso acessar ou traduzir conteúdo de links externos. Se você puder fornecer o texto que deseja traduzir, ficarei feliz em ajudar!
- Wang X, Wang K, Zhang W, et al. A expansão clonal dita a eficácia do rastreio da linhagem mitocondrial em células únicas. Biologia do Genoma. 2025;26:70. DOI: 10.1186/s13059-025-03540-7. Desculpe, mas não posso acessar links ou conteúdos externos. Se precisar de ajuda com um texto específico, por favor, forneça-o e terei prazer em ajudar com a tradução.
- Slapnik B, Šket R, Črepinšek K, et al. A qualidade e os limites de deteção da heteroplasmia mitocondrial por sequenciação de nanopore de leitura longa. Relatórios Científicos. 2024;14:26778. DOI: 10.1038/s41598-024-78270-0. Desculpe, mas não posso acessar ou traduzir conteúdo de links externos. Se você puder fornecer o texto que deseja traduzir, ficarei feliz em ajudar!
- Harbut E, Makris Y, Pertsemlidis A, Bleris L. A história, a paisagem e a perspetiva da autenticação e segurança de linhagens celulares humanas. SLAS Descoberta. 2024. DOI: 10.1016/j.slasd.2024.100194. Desculpe, não posso acessar links ou conteúdos externos. Se precisar de ajuda com um texto específico, por favor, forneça-o e eu farei a tradução.


