
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
Análise Comparativa de Sequências de mtDNA: Decifrando a Divergência Funcional Mitocondrial em Modelos de Investigação
A comparação de sequências mitocondriais torna-se difícil não quando as variantes são difíceis de detectar, mas quando a saída se torna demasiado fácil de sobreinterpretar. Em muitos projetos RUO, as equipas já têm leituras brutas aceitáveis, uma lista de variantes e uma tabela de heteroplasmia, no entanto, a próxima questão permanece sem resposta: o que a comparação das sequências de DNA mitocondrial realmente mostrou? Requer uma comparação estruturada entre regiões genómicas, classe de mutação, nível de heteroplasmia, contexto de conservação e qualidade de reporte. O resultado deve ser mais do que uma lista de diferenças; deve ser uma estrutura de interpretação classificada que permaneça compatível com a análise subsequente e a revisão de entregas de fornecedores. A análise de mtDNA também tem várias limitações técnicas bem conhecidas, incluindo a escolha da sequência de referência, a interferência de NUMT e a sensibilidade do limiar para heteroplasmia de baixa frequência, portanto, a interpretação biológica deve sempre estar ligada ao controlo de qualidade e às suposições do pipeline.
Comparado com fluxos de trabalho genómicos mais amplos, a comparação do mtDNA tem um alvo genómico mais restrito, mas uma carga de interpretação mais subtil. O genoma mitocondrial é compacto, denso em genes e funcionalmente acoplado à fosforilação oxidativa, por isso um pequeno número de diferenças bem classificadas pode ser mais informativo do que uma longa lista não ordenada. Ao mesmo tempo, nem toda diferença é funcionalmente significativa. As alterações na região codificadora precisam de ser separadas da variação na região de controlo, as substituições sinónimas das candidatas não sinónimas, e os padrões estáveis específicos de grupo dos sinais de baixo nível ruidosos. Para projetos de investigação B2B, essa distinção é importante porque o verdadeiro resultado não é apenas dados de sequência, mas uma narrativa analítica pronta para decisão: quais variantes merecem seguimento, quais diferenças podem refletir o fundo de linhagem em vez de divergência funcional, e quais sinais permanecem provisórios devido a preocupações com profundidade, referência ou contaminação.
A Questão Central: O Que Mostrou a Comparação das Sequências de DNA Mitocondrial?
No nível mais básico, a análise comparativa de sequências de mtDNA deve responder a quatro questões: onde estão localizadas as diferenças, que tipo de alterações de sequência são, com que frequência ocorrem dentro e entre grupos, e se o padrão observado é plausível como uma hipótese funcional em vez de ser apenas um marcador de linhagem. Uma boa comparação começa, portanto, por dividir o genoma em loci codificadores de proteínas, loci de rRNA/tRNA, e o D-loop/região de controloAs alterações na região codificadora geralmente impulsionam a primeira ronda de priorização funcional, especialmente para genes que contribuem para os complexos OXPHOS, enquanto as diferenças na D-loop são mais frequentemente interpretadas no contexto da regulação, sinais associados à replicação ou discriminação de grupos, em vez de um efeito imediato a nível de proteína. Essa distinção evita um erro comum e precoce: tratar todas as diferenças no mtDNA como equivalentes.
A segunda camada é a classe de mutações. Uma estrutura de revisão útil separa alterações sinónimas, substituições não sinónimas, pequenas indels e variantes não codificantes. Na prática, a lista de candidatos da primeira passagem geralmente prioriza substituições não sinónimas em posições conservadas, seguidas por alterações não codificantes recorrentes que estão fortemente associadas a grupos e são tecnicamente robustas. Isso não significa que as alterações sinónimas ou da região de controlo devam ser ignoradas; significa que não devem ocupar o mesmo nível interpretativo que os candidatos que alteram aminoácidos até que evidências adicionais estejam disponíveis. Uma forma prática de apresentar isso num relatório é anotar cada local com contexto genómico, consequência do aminoácido, profundidade, estimativa de heteroplasmia e uma simples bandeira de priorização. Esse formato é muito mais fácil para um responsável pela análise subsequente rever do que um excerto bruto de VCF sozinho.
A terceira camada é a heteroplasmia. Em projetos comparativos, o mesmo locus pode comportar-se de forma muito diferente dependendo de se o alelo alternativo é quase homoplasmático, moderadamente heteroplasmático ou observado apenas numa baixa fração. A heteroplasmia não é uma métrica decorativa; altera a confiança com que se deve interpretar as diferenças entre grupos. Um sítio presente a alta profundidade e com heteroplasmia reprodutível entre réplicas é analiticamente diferente de uma chamada de baixa frequência na borda do limite de deteção. Avaliações recentes mostram que a sensibilidade e fiabilidade da deteção de heteroplasmia dependem fortemente da profundidade, tecnologia e perfil de erro, especialmente quando a análise tenta resolver misturas de baixo nível. Por esta razão, a interpretação entre grupos é mais forte quando as distribuições de heteroplasmia são comparadas juntamente com a profundidade de leitura, equilíbrio de fita e reprodutibilidade, e não como percentagens isoladas.
 Figura 1. Paisagem Comparativa de Variantes de mtDNA entre Grupos de Pesquisa.
Figura 1. Paisagem Comparativa de Variantes de mtDNA entre Grupos de Pesquisa.
Um mapa circular de mtDNA destacando regiões codificadoras, o D-loop, a densidade de hotspots específicos de grupo e a intensidade de heteroplasmia, para que os leitores possam distinguir locais candidatos de alta prioridade da variação de fundo.
Uma regra prática de revisão é esta: a comparação deve mostrar padrão, não apenas presençaSe a saída apenas prova que existem variantes, a análise está incompleta. Se mostrar que certos loci estão enriquecidos em um grupo de pesquisa, que as alterações de codificação candidatas se agrupam em regiões funcionalmente relevantes e que os padrões de heteroplasmia estão consistentemente deslocados entre os grupos, então a comparação começa a apoiar uma hipótese de divergência funcional adequada para seguimento em RUO. O objetivo não é reivindicar o mecanismo muito cedo, mas estabelecer um caminho classificado da diferença de sequência à questão experimental.
Fluxo de Trabalho para Análise Avançada de Sequências de DNA Mitocondrial
Um fluxo de trabalho avançado de comparação de mtDNA geralmente começa com a revisão padronizada de entradas. Para a avaliação de entregas de fornecedores, isso significa verificar se o pacote do projeto inclui arquivos FASTQ limpos, arquivos BAM prontos para alinhamento ou pré-alinhados, um arquivo de variantes ou conjunto de chamadas tabular, e um resumo de QC suficientemente detalhado. Para uma revisão ao estilo M-02, apenas o Q30 não é suficiente; o pacote também deve esclarecer a profundidade média de mtDNA, a fração no alvo se foi utilizada enriquecimento, o comportamento de duplicação e a lógica de cálculo da heteroplasmia. A compatibilidade do pipeline melhora substancialmente quando as convenções de nomenclatura, a construção de referência e o esquema de anotação são explícitos desde o início. É também aqui que a qualidade do método upstream importa: a interpretação comparativa de alta confiança depende da geração de dados adequada ao propósito, seja o estudo construído em torno de fluxos de trabalho de sequenciação de mtDNA ou mais direcionado sequenciação de amplicõesOs leitores que trabalham retrocedendo a partir de problemas de análise também devem rever Otimização do protocolo de sequenciação de mtDNA para amostras complexas antes de atribuir padrões ambíguos a jusante à biologia em vez de ao design a montante.
Após a revisão de entrada, o alinhamento de múltiplas sequências e o manuseio de referências tornam-se o próximo ponto de controlo. Para comparações focadas no ser humano, a Sequência de Referência de Cambridge revista continua a ser uma base comum para a reportação de posições e harmonização de anotações, e o MITOMAP continua a funcionar como um recurso interpretativo atual para pesquisa de lócus, busca de alelos, análise de sequências baseada no MITOMASTER e contexto de posição ligado ao rCRS. Em modelos RUO relevantes para humanos, a consistência com coordenadas baseadas no rCRS apoia uma comparação mais clara entre estudos. Em projetos não humanos ou interespécies, no entanto, a portabilidade de coordenadas por si só não é suficiente; a análise deve separar explicitamente a "conveniência de referência humana" da "comparação biológica apropriada à espécie." Isso geralmente significa alinhar primeiro dentro das espécies, e depois usar uma interpretação consciente da conservação para inferência funcional entre espécies.
Para pipelines automatizados ou semi-automatizados, ferramentas como o MToolBox são valiosas porque fazem mais do que apenas montar ou chamar variantes. Elas também suportam saídas conscientes da heteroplasmia e lógica de priorização, o que é útil quando o revisor precisa de mais do que uma lista plana de locais. Um fluxo de trabalho robusto normalmente inclui QC de leitura, alinhamento ou montagem focada em mtDNA, filtragem consciente de NUMTs, avaliação de cobertura, chamada de variantes consciente de heteroplasmia, anotação e uma camada final de priorização. Os NUMTs merecem atenção especial aqui. Fragmentos mitocondriais embutidos no núcleo podem distorcer o sinal aparente de mtDNA se o fluxo de trabalho não os controlar nem no design do laboratório molhado nem no filtro computacional. Em termos práticos, qualquer sinal de baixa fração inesperadamente difuso, especialmente se repetido em amostras não relacionadas, deve acionar uma verificação de NUMT antes que a interpretação biológica se expanda. Onde a reutilização a montante é importante, uma estrutura organizada pipeline de chamada de variantes e compatível sequenciação de biblioteca pré-fabricada as estratégias são úteis apenas quando as suposições de referência, filtragem e anotação são suficientemente transparentes para reutilização interna.
A previsão funcional é melhor aplicada como uma camada de priorização, não como um substituto para a evidência. Para candidatos não sinónimos em genes codificadores de proteínas do mtDNA, ferramentas como SIFT e PolyPhen-2 podem ajudar a classificar as substituições pelo provável impacto estrutural ou funcional. Isso é especialmente útil quando a comparação gera muitas alterações de aminoácidos e a equipa precisa de uma lista curta para uma revisão mais aprofundada. No entanto, essas ferramentas devem ser usadas para triagem comparativa em vez de inflacionar conclusões: a mudança em locais conservados mais a pontuação de dano prevista mais o enriquecimento consistente do grupo resulta num candidato de seguimento mais forte, não numa declaração final de mecanismo.
Interpretando a Divergência Funcional em Contextos de Pesquisa B2B
Em modelos de pesquisa orientados para o metabolismo, uma sequência interpretativa útil é: detectar variantes específicas de grupo, priorizar pelo contexto genómico e conservação, estimar a provável consequência proteica e, em seguida, conectar os candidatos mais fortes a resultados de pesquisa mensuráveis, como eficiência associada ao ATP, equilíbrio redox ou diferenças na resposta ao stress. A redação é importante. Em ambientes de RUO, a conclusão correta é geralmente "apoia uma hipótese de comportamento funcional mitocondrial alterado" em vez de "prova um efeito definido." As equipas que mais tarde desejam confirmação ortogonal podem estender a comparação com quantificação do número de cópias de mtDNA ou com um mais amplo fluxo de integração multi-ômica, dependendo do âmbito do projeto.
A comparação entre espécies acrescenta outro filtro interpretativo: a conservação. Uma posição que é estável entre organismos relacionados, mas alterada em um modelo, pode merecer mais atenção do que uma mudança em um local naturalmente variável. É aqui que muitos artigos se tornam demasiado centrados no ser humano. Se o projeto compara modelos não humanos, a análise não deve depender apenas da conveniência das coordenadas humanas. Em vez disso, deve integrar um alinhamento consciente das espécies e uma pontuação de conservação, e deve sinalizar claramente quando uma conclusão é apoiada pela conservação entre espécies em vez de recursos de anotação apenas humanos. Essa distinção é frequentemente a diferença entre um relatório comparativo persuasivo e um que é difícil de defender em uma revisão técnica.
Uma matriz de priorização prática para a divergência funcional geralmente inclui região genómica, classe de mutação, intervalo de heteroplasmia, contexto de conservação, impacto proteico previsto, se aplicável, enriquecimento entre grupos e próximo passo recomendado.
Para tornar a interpretação comparativa operacional, o relatório deve passar de uma prosa descritiva para uma matriz de priorização fixa. Em ambientes RUO, cada local candidato deve ser revisto nos mesmos campos: região genómica, classe de mutação, comportamento de heteroplasmia, contexto de conservação, confiança técnica, enriquecimento entre grupos e ação recomendada a seguir. Isso previne o problema comum de sobrevalorizar variantes visualmente impressionantes que carecem de reprodutibilidade ou contexto. Também torna os entregáveis externalizados mais fáceis de comparar com os pipelines internos, uma vez que a lógica de classificação se torna explícita em vez de dependente do revisor. Para a maioria dos projetos, uma saída de três níveis é suficiente: candidatos de acompanhamento de alta prioridade, diferenças contextuais mas de menor prioridade, e chamadas provisórias que requerem revisão de QC antes que a interpretação se expanda.
Matriz de Priorização Pronta para Decisão
| Site/Região | Classe de Mutação | Padrão de Heteroplasmia | Conservação | Confiança Técnica | Enriquecimento de Grupo | Camada de Prioridade | Próximo Passo RUO Recomendado |
|---|---|---|---|---|---|---|---|
| Locus codificante conservado | Não sinónimo | Deslocamento reproduzível entre réplicas | Alto | Alta profundidade, suporte equilibrado | Forte | Alto | Revisão do impacto das proteínas mais acompanhamento ortogonal direcionado |
| Locus de codificação | Sinónimo | Estável mas modesto | Moderado | Alto | Moderado | Médio | Mantenha como sinal contextual; não conduza a interpretação sozinha. |
| ponto quente do D-loop | Não codificante | Sinal específico de grupo forte | Variável | Alto | Forte | Médio | Tratar como candidato associado a regulamentos ou linhagens; validar a consistência. |
| Qualquer região | Qualquer aula | Perto do limiar ou instável | Qualquer | Baixa profundidade ou suporte inconsistente | Pouco claro | Provisório | Rever QC, filtragem e contexto da amostra antes da interpretação. |
| Chamadas de baixo nível recorrentes entre amostras não relacionadas | Qualquer aula | Difundir | Qualquer | Suspeito | Fraco | Provisório | Revise o manuseio de NUMT e a especificidade de alinhamento primeiro. |
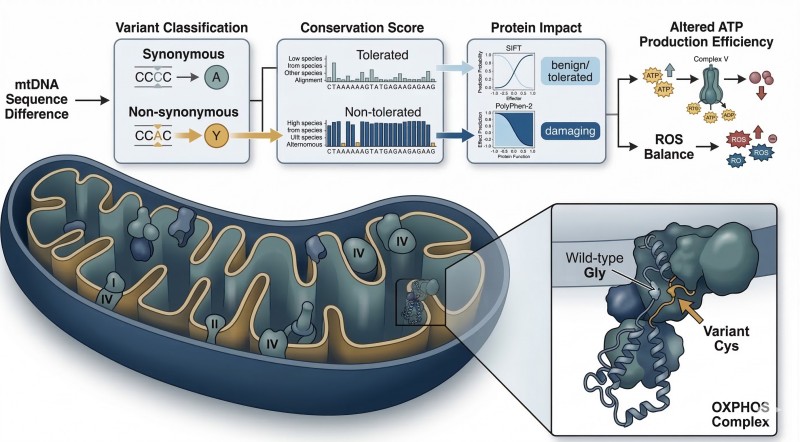 Figura 2. Das Diferenças de Sequência do mtDNA às Hipóteses de Divergência Funcional.
Figura 2. Das Diferenças de Sequência do mtDNA às Hipóteses de Divergência Funcional.
Um mapa lógico em camadas que liga diferenças de sequência à previsão de impacto proteico e hipóteses a nível de via envolvendo o comportamento do OXPHOS, a produção associada ao ATP e observações de pesquisa relacionadas com ROS.
Uma forma útil de apresentar o resto da interpretação é como um quadro de "o que observar a seguir".
Categorias comuns de características mitocondriais e pontos de observação em pesquisa
Genes OXPHOS que codificam proteínas
Priorize substituições não sinónimas, especialmente em resíduos conservados. Observe ensaios ligados ao ATP, saídas associadas ao redox ou leituras relacionadas ao potencial de membrana no sistema modelo.
loci de rRNA e tRNA
Interprete com cautela, mas não ignore. Estes podem influenciar a eficiência mitocondrial relacionada com a tradução e merecem uma anotação estruturada em vez de um desprezo padrão. O MITOMAP e o MITOMASTER continuam a ser úteis para o contexto da posição da sequência, enquanto recursos de anotação expandidos melhoraram o suporte à interpretação de mt-tRNA.
D-loop / região de controlo
Melhor avaliado para controlo de replicação, contexto regulatório e separação de grupos, especialmente quando repetido em amostras com forte suporte técnico. A sobre-interpretação é comum aqui, a menos que a diferença seja estável e claramente enriquecida.
Superando Gargalos de Análise: De Listas de Variantes a Insights Biológicos
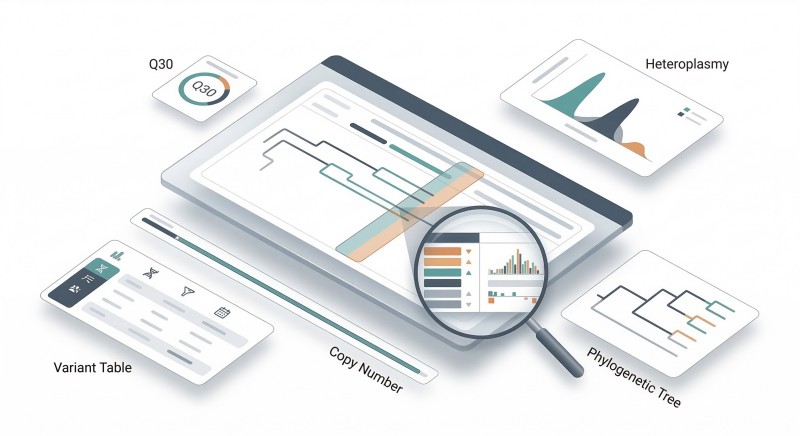 Figura 3. Relatório de Análise Comparativa de mtDNA: Desde Métricas de QC até Priorização Biológica.
Figura 3. Relatório de Análise Comparativa de mtDNA: Desde Métricas de QC até Priorização Biológica.
Um painel de estilo relatório que mostra a combinação de métricas de QC, distribuição de heteroplasmia, agrupamento filogenético, anotação de variantes e contexto de número de cópias necessário para uma análise comparativa pronta para decisão.
O gargalo mais comum no trabalho comparativo de mtDNA não é a deteção de variantes. É a lacuna entre um conjunto de chamadas tecnicamente aceitável e uma lista curta biologicamente útil. Quando o relatório contém dezenas ou centenas de diferenças, as equipas muitas vezes precisam de uma segunda camada interpretativa que combine a variação de sequência com o contexto da amostra e a confiança analítica. É por isso que os melhores relatórios comparativos são de múltiplos painéis em vez de saídas de tabela única. Eles devem mostrar um resumo compacto de QC, uma visão da distribuição de heteroplasmia, uma tabela de variantes candidatas agrupadas e um resumo consciente da linhagem que ajuda a distinguir a estrutura de ancestralidade de fundo dos sinais de prioridade funcional. Antes de estender a interpretação, as equipas devem confirmar a identidade da amostra através de identificação de linhagens celulares e rever o contexto de linhagem relacionado através de rastreio da linhagem mitocondrial na autenticação de linhas celulares RUO.
Outro estrangulamento é a fraca relação entre a contagem de variantes e a perspetiva biológica. Mais variantes não significam necessariamente mais valor explicativo. Em muitos projetos, a interpretação torna-se mais clara quando as diferenças na sequência do mtDNA são analisadas em conjunto com o contexto do número de cópias. O número de cópias de mtDNA não substitui a comparação de sequências, mas pode adicionar uma segunda dimensão útil quando a equipa quer saber se um modelo difere apenas pela composição da sequência ou também pela abundância genómica mitocondrial. Mesmo na literatura de investigação não clínica, as análises do número de cópias de mtDNA demonstraram que a abundância genómica pode alterar materialmente a forma como as hipóteses sobre o estado mitocondrial são formuladas.
QC e resolução de problemas: sintoma → causa provável → próxima ação
Números inesperadamente elevados de variantes de baixa frequência em muitas amostras
Causa provável: interferência de NUMT, ambiguidade de mapeamento ou limiares de chamada excessivamente permissivos.
Próxima ação: inspecionar a especificidade de alinhamento, rever a estratégia de enriquecimento de mtDNA, apertar os filtros do chamador e rever as regiões conhecidas sensíveis a NUMT.
As estimativas de heteroplasmia variam drasticamente entre repetições técnicas.
Causa provável: profundidade insuficiente, comportamento de erro específico da plataforma ou pré-processamento inconsistente.
Próxima ação: verificar a distribuição de profundidade, suporte de filamento, manuseio de duplicados e a definição exata de heteroplasmia utilizada no relatório. O estudo de referência de leituras longas de 2024 relatou um limiar de deteção de heteroplasmia de 12% a 150× de cobertura no seu ambiente avaliado, o que é um lembrete útil de que os limites de deteção dependem da tecnologia e do fluxo de trabalho, em vez de serem universais.
Muitas diferenças, mas pouca estrutura de grupo interpretável.
Causa provável: mistura de linhagens de fundo, inconsistência na identidade da amostra ou variantes de baixa prioridade a dominarem o relatório.
Próxima ação: aplicar agrupamento consciente da linhagem, priorizar candidatos de codificação conservada e confirmar a consistência da amostra antes de estender a interpretação funcional.
Sinal forte do D-loop, mas sinal fraco na região codificadora.
Causa provável: enriquecimento de regiões regulatórias, separação de linhagens ou variação com forte influência na região de controlo sem consequência direta para a proteína.
Próxima ação: evitar forçar uma narrativa de impacto proteico; em vez disso, posicionar o resultado como uma descoberta regulatória ou de comparação, a menos que outras evidências a fortaleçam.
Bom Q30, mas fraca interpretabilidade a montante.
Causa provável: entregas incompletas, anotação superficial ou relatórios não estruturados.
Próxima ação: solicitar dados brutos e saídas de anotação padronizados, informações de referência explícitas e uma tabela de variantes pronta para priorização. Uma boa qualidade base não compensa uma embalagem interpretativa deficiente.
Conclusão: Reforçar os Estudos RUO com Análises Comparativas Precisas
A análise comparativa de sequências de mtDNA é mais útil quando transforma a diferença de sequência em um caminho de interpretação classificado e tecnicamente defensável. Para modelos de pesquisa RUO, isso significa olhar além da presença bruta de variantes e perguntar onde a diferença se encontra, que classe de mudança representa, quão robusto é o sinal de heteroplasmia, se o local é conservado e quão facilmente o resultado pode ser integrado em raciocínios biológicos subsequentes. Uma comparação bem conduzida não colapsa tudo em um único rótulo "funcional". Em vez disso, separa a variação de fundo provável, o sinal associado a linhagens e candidatos que merecem acompanhamento. Essa abordagem é especialmente valiosa para equipas de pesquisa B2B que necessitam de rigor analítico e compatibilidade de pipeline nas entregas dos fornecedores.
Olhando para o futuro, as estratégias de leitura longa podem melhorar ainda mais a interpretação de moléculas completas, o contexto de fase e o comportamento de deteção em cenários difíceis de heteroplasmia, especialmente quando o projeto precisa de mais confiança em sinais complexos ou de baixo nível. A recente avaliação comparativa sugere que as abordagens de leitura longa são promissoras, mas o seu comportamento analítico ainda precisa ser compreendido no contexto dos limites de deteção e das características de erro, em vez de se assumir que são superiores em todos os casos de uso. Em outras palavras, uma melhor análise comparativa virá não apenas de mais sequenciação, mas de uma melhor classificação de evidências, entregáveis mais claros e uma ligação mais estreita entre o controlo de qualidade e a interpretação.
Perguntas Frequentes
1) O que a comparação das sequências de ADN mitocondrial mostrou num projeto típico de RUO?
Geralmente, mostra se as diferenças estão concentradas em regiões codificadoras, regiões de controlo ou compartimentos genómicos mistos; se as alterações são sinónimas ou não sinónimas; e se as distribuições de heteroplasmia diferem entre grupos de investigação de uma forma tecnicamente consistente. O resultado mais forte é um conjunto priorizado de candidatos, e não apenas uma lista de variantes mais longa.
2) A sequência do DNA mitocondrial humano é sempre a referência certa para comparação?
Não. Projetos orientados para humanos frequentemente beneficiam de relatórios baseados em rCRS para consistência, mas modelos interespécies ou não humanos necessitam de comparação consciente das espécies e interpretação baseada na conservação, em vez de se basearem apenas na conveniência de um referência humana.
3) Como deve um responsável de bioinformática rever um relatório de comparação de mtDNA subcontratado?
Comece com a completude dos entregáveis: saída de alinhamento FASTQ/BAM ou equivalente, tabela de variantes anotadas, estimativas de heteroplasmia, detalhes de referência e métricas de QC claras, incluindo profundidade. Em seguida, reveja a lógica de classificação de candidatos e se o relatório separa a variação de fundo dos locais de prioridade funcional.
4) É Q30 suficiente para avaliar a qualidade dos dados para comparação de mtDNA?
Não. A Q30 é útil, mas incompleta. A profundidade média do mtDNA, a distribuição das leituras, o suporte à heteroplasmia e as suposições de filtragem são todos importantes para a interpretabilidade, especialmente quando sinais de baixa frequência estão a ser comparados entre grupos.
5) Podem as diferenças de heteroplasmia ser interpretadas diretamente como divergência funcional?
Não por si só. A heteroplasmia suporta a interpretação quando é tecnicamente robusta e alinhada com o contexto genómico, conservação e relevância do local candidato. É uma camada na cadeia de inferência, não a cadeia inteira.
6) Por que é que os NUMTs são importantes na análise de sequências mitocondriais?
Porque fragmentos mitocondriais incorporados no núcleo podem imitar o sinal do mtDNA e criar chamadas de fração baixa enganosas ou artefatos recorrentes se a filtragem for fraca. O controlo de NUMT é importante tanto no desenho experimental como na análise de dados.
7) Quando deve o número de cópias de mtDNA ser adicionado a um estudo de comparação?
Quando o projeto necessita de uma segunda dimensão além da composição da sequência, especialmente se a equipa quiser comparar a abundância genómica mitocondrial juntamente com a carga de variantes ou o padrão de heteroplasmia. É mais útil como contexto complementar, não como um substituto para a análise de sequências.
8) O que torna um relatório de mtDNA compatível com pipelines?
Sistema de coordenadas consistente, sequência de referência explícita, formatos de ficheiro padrão, campos de anotação reproduzíveis e uma estrutura de priorização que pode ser reintegrada em fluxos de trabalho subsequentes sem reconstrução manual.
Referências
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanálise e revisão da sequência de referência de Cambridge para o DNA mitocondrial humano. Genética da Natureza. 1999;23(2):147. DOI: 10.1038/13779.
- Wallace DC, Lott MT, Brown MD, et al. MITOMAP: Uma Base de Dados do Genoma Mitocondrial Humano. Pesquisa em Ácidos Nucleicos. 1996;24(1):177-179. DOI: 10.1093/nar/24.1.177.
- MITOMAP. MITOMAP: Uma Base de Dados do Genoma Mitocondrial Humano. Formato de citação do site MITOMAP, 2023; as ferramentas atuais incluem Pesquisa de Alelos e MITOMASTER, com o site a mostrar atualizações recentes da base de dados. Recurso MITOMAP.
- Calabrese C, Simone D, Diroma MA, et al. MToolBox: uma pipeline altamente automatizada para anotação de heteroplasmia e análise de priorização de variantes mitocondriais humanas em sequenciação de alto débito. Bioinformática. 2014;30(21):3115-3117. DOI: 10.1093/bioinformatics/btu483.
- Weissensteiner H, Pacher D, Kloss-Brandstätter A, et al. Uma coleção abrangente de anotações para interpretar a variação de sequência em transfer RNAs mitocondriais humanos. BMC Bioinformática. 2016;17:420. DOI: 10.1186/s12859-016-1193-4.
- Adzhubei IA, Schmidt S, Peshkin L, et al. Um método e servidor para prever mutações missense prejudiciais. Métodos da Natureza. 2010;7(4):248-249. DOI: 10.1038/nmeth0410-248.
- Ng PC, Henikoff S. SIFT: Prever alterações de aminoácidos que afetam a função da proteína. Pesquisa em Ácidos Nucleicos. 2003;31(13):3812-3814. DOI: 10.1093/nar/gkg509.
- Sturk-Andreaggi K, Renshaw M, et al. Heteroplasmia do mtDNA: Origem, Detecção, Significado e Consequências Evolutivas. Vida. 2021;11(7):633. DOI: 10.3390/life11070633.
- Dayama G, Emery SB, Kidd JM, Mills RE. A paisagem genómica de inserções mitocondriais nucleares polimórficas humanas. Pesquisa em Ácidos Nucleicos. 2014;42(20):12640-12649. DOI: 10.1093/nar/gku1038.
- Slapnik B, Šket R, Črepinšek K, Tesovnik T, Jenko Bizjan B, Kovač J. A qualidade e os limites de deteção da heteroplasmia mitocondrial através da sequenciação por nanopore de leitura longa. Relatórios Científicos. 2024;14:26778. DOI: 10.1038/s41598-024-78270-0.
- Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Pensar fora do núcleo: Número de cópias de ADN mitocondrial na saúde e na doença. Mitocôndria. 2020;53:214-223. DOI: 10.1016/j.mito.2020.06.004. (Europe PMC)


