
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
Um Guia Atualizado para a Preparação de Bibliotecas Illumina: Kits, Métodos e Estratégias para Tipos de Amostras Desafiadoras
A preparação da biblioteca é o único fator mais importante para o sucesso do sequenciamento. Independentemente de quão poderosa seja a plataforma de sequenciamento ou quão sofisticado seja o pipeline de análise de dados, uma biblioteca mal preparada produzirá dados inutilizáveis, desperdício de capacidade do fluxo celular e perda de tempo.
Mas em 2026, o desafio para a maioria dos investigadores já não é compreender os passos básicos da preparação de bibliotecas. O desafio é escolher o método certo entre um campo de opções cada vez mais saturado—baseado em PCR vs. livre de PCR, baseado em fragmentação vs. baseado em tagmentação, e kits otimizados para tipos de amostras específicas, como tecidos FFPE, ADN livre circulante (cfDNA) e amostras de ultra-baixo input.
Este guia fornece uma estrutura prática para tomar essas decisões. Abrange as principais estratégias de preparação de bibliotecas e kits disponíveis para sequenciação Illumina, com foco na seleção da abordagem certa para o seu tipo de amostra, escala do projeto e requisitos de qualidade dos dados.
Por que as Escolhas de Preparação de Bibliotecas Importam Mais do Que Nunca
A preparação de bibliotecas representa a maior parte da variabilidade nos fluxos de trabalho de NGS. Uma biblioteca bem concebida produz consistentemente dados de alta qualidade; uma mal concebida pode falhar mesmo no melhor instrumento de sequenciação. À medida que os custos de sequenciação diminuíram, a preparação de bibliotecas tornou-se uma parte proporcionalmente maior do custo total do projeto, tornando a escolha do método uma decisão financeira significativa.
Três fatores chave influenciam a escolha do método de preparação da biblioteca:
- Qualidade e quantidade da amostraO DNA genómico de alta qualidade (>1 µg) permite o acesso a métodos sem PCR que minimizam o viés. Amostras de baixa qualidade ou baixa quantidade (FFPE, cfDNA, células únicas) requerem kits especializados com enzimas de reparo ou protocolos de ultra-baixa entrada. A causa mais comum de falha na biblioteca é o material de entrada degradado que não foi identificado até após a quantificação da biblioteca — razão pela qual investir em um controlo de qualidade rigoroso desde o início compensa muitas vezes.
- Tipo de projetoO sequenciamento de genoma completo (WGS) exige baixo viés experimental e cobertura consistente. O sequenciamento direcionado requer alta eficiência de captura. O sequenciamento por amplicão depende de primers multiplex equilibrados. Cada tipo de aplicação impõe diferentes exigências ao método de preparação da biblioteca, o que significa que não existe um único "melhor" kit—apenas o melhor kit para uma dada combinação de amostra e aplicação.
- Rendimento e tempo de respostaAlguns métodos podem passar de DNA para uma biblioteca pronta para sequenciação em menos de 3 horas; outros requerem 6 a 8 horas. Para grandes lotes, a compatibilidade com a automação torna-se um fator importante. Um laboratório que processa 96 amostras por semana terá requisitos de rendimento diferentes de um que processa 16 amostras por mês.
Além destes fatores, uma quarta consideração está a ganhar importância: a complexidade da biblioteca. Uma biblioteca com alta complexidade—o que significa que representa o genoma ou transcriptoma original com mínima duplicação—produz chamadas de variantes de maior qualidade, medições de expressão mais fiáveis e resultados mais reproduzíveis entre lotes. Métodos de preparação de bibliotecas que preservam a complexidade (sem PCR, PCR de baixo ciclo e protocolos de tagmentação otimizados) estão a ser cada vez mais favorecidos para projetos onde a qualidade dos dados é a principal preocupação.
Para investigadores que estão a planear o seu primeiro projeto de NGS ou que procuram otimizar um pipeline existente, compreender estas compensações é essencial. Abrangente serviços de NGS cobrir toda a gama de métodos de preparação de bibliotecas, tornando possível selecionar a abordagem ideal para os requisitos específicos de cada projeto.
O Fluxo de Trabalho Padrão em Resumo
Os passos principais da preparação de bibliotecas da Illumina permanecem consistentes na maioria dos métodos, embora a implementação específica varie:
- FragmentaçãoO DNA é fragmentado em fragmentos de tamanho alvo (tipicamente 200–800 pb) por cisalhamento mecânico, digestão enzimática ou tagmentação.
- Reparação de extremidades e A-tailingAs extremidades dos fragmentos são embotadas, fosforiladas e com cauda A para permitir a ligação do adaptador.
- Ligação de adaptadoresAdaptadores de sequenciação contendo sequências P5/P7, códigos de barras de índice e locais de ligação de primers de sequenciação são ligados aos fragmentos.
- Seleção de tamanhoFragmentos fora da faixa de tamanho alvo são removidos, tipicamente utilizando esferas magnéticas SPRI.
- Amplificação da biblioteca (opcional)A amplificação por PCR adiciona material suficiente para sequenciação. Os métodos sem PCR saltam esta etapa completamente.
- QC da BibliotecaA biblioteca final é quantificada e verificada em termos de qualidade utilizando qPCR, ensaios fluorimétricos e eletroforese capilar (Bioanalyzer ou TapeStation).
Esta visão geral é intencionalmente breve porque a mecânica detalhada de cada passo já está coberta em recursos existentes. O foco deste guia é na escolha entre os métodos disponíveis, e não no protocolo passo a passo.
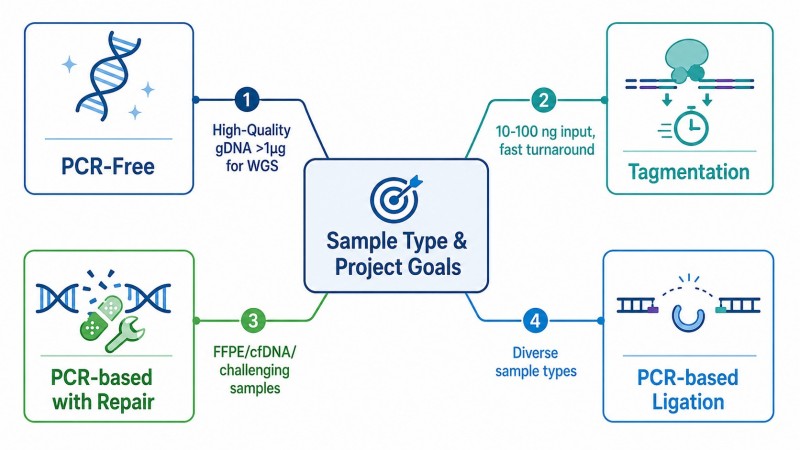 Figura 1. Estrutura de decisão para preparação de bibliotecas — do tipo de amostra e objetivos do projeto à estratégia de preparação ideal
Figura 1. Estrutura de decisão para preparação de bibliotecas — do tipo de amostra e objetivos do projeto à estratégia de preparação ideal
Estrutura de decisão que orienta os investigadores desde o tipo de amostra, quantidade de entrada e objetivos do projeto até à estratégia de preparação de biblioteca ideal, abrangendo métodos baseados em PCR, sem PCR e de tagmentação.
Escolhendo a Estratégia de Preparação de Biblioteca Certa — Baseada em PCR, Sem PCR ou Tagmentação
Cada método de preparação de bibliotecas da Illumina se enquadra em uma das três categorias técnicas. Compreender as diferenças entre estas categorias é o primeiro passo para selecionar o kit certo para o seu projeto.
Fragmentação baseada em PCR + ligaduraO DNA é fragmentado por cisalhamento mecânico (Covaris) ou digestão enzimática, seguido de reparação das extremidades, adição de caudas A, ligação de adaptadores e amplificação por PCR. Esta é a abordagem mais flexível e amplamente utilizada. Funciona bem numa ampla gama de quantidades de entrada (0,1 ng a 1 µg) e tipos de amostras. A desvantagem é que a amplificação por PCR pode introduzir viés em regiões ricas em GC e aumentar as taxas de duplicação. Os kits que utilizam esta estratégia normalmente requerem de 4 a 8 horas para um fluxo de trabalho completo.
Fragmentação sem PCR + ligaduraO mesmo fluxo de trabalho que o acima, mas a amplificação por PCR é omitida. Isso elimina o viés introduzido pela PCR e produz a cobertura do genoma mais uniforme, tornando-se o padrão ouro para aplicações de WGS. A limitação é que os métodos sem PCR requerem uma maior quantidade de DNA de entrada (tipicamente >100 ng a >1 µg, dependendo do kit), uma vez que não há etapa de amplificação para aumentar o rendimento da biblioteca.
Tagmentação (baseada em transposase)Uma enzima transposase modificada fragmenta simultaneamente o DNA e insere sequências de adaptadores em um único passo de reação. Isso reduz significativamente o tempo de manuseio e os requisitos de entrada — alguns kits de tagmentação funcionam com apenas 1 ng de DNA de entrada. A desvantagem é que os métodos de tagmentação podem introduzir viés dependente da sequência, particularmente em regiões de baixo GC, e podem produzir bibliotecas com uma distribuição de tamanhos de inserção mais estreita.
Quadro de seleçãoCom base na análise acima, um guia prático para a seleção de métodos pode ser resumido da seguinte forma:
- DNA de alta qualidade >1 µg para WGS → Fragmentação sem PCR + ligadura produz a cobertura mais uniforme com as taxas de duplicação mais baixas
- Entrada de 10–100 ng, necessidade de resposta rápida → Baseada em tagmentação os métodos são os mais eficientes, reduzindo o tempo de preparação da biblioteca em cerca de metade
- FFPE, cfDNA ou outras amostras desafiantes → Fragmentação baseada em PCR + ligação com enzimas de reparo ou kits especializados de ultra-baixo input
- Laboratório flexível e multifuncional a processar diversos tipos de amostras. Fragmentação e ligação baseadas em PCR oferece a mais ampla gama de entrada e compatibilidade de aplicação
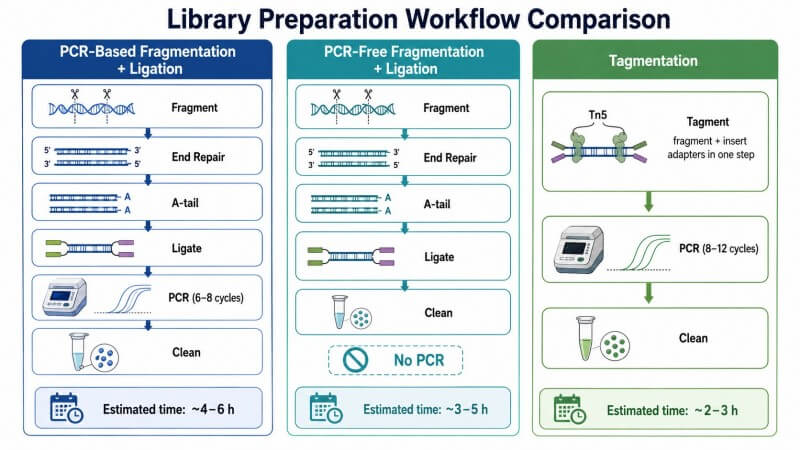 Figura 2. Três estratégias de preparação de bibliotecas — fragmentação baseada em PCR + ligadura, sem PCR e tagmentação
Figura 2. Três estratégias de preparação de bibliotecas — fragmentação baseada em PCR + ligadura, sem PCR e tagmentação
Legenda: Comparação das três principais estratégias de preparação de bibliotecas Illumina, mostrando as diferenças no mecanismo de fragmentação, requisitos de amplificação, intervalo de DNA de entrada e aplicações mais adequadas para cada abordagem.
Considerações sobre o fluxo de trabalho no laboratórioAlém dos fatores técnicos acima, a logística prática de laboratório deve informar a escolha. Os kits de fragmentação + ligadura baseados em PCR produzem resultados consistentes numa ampla variedade de tipos de amostras, tornando-os ideais para instalações centrais ou laboratórios de serviço que recebem tipos de amostras diversos. Os métodos de tagmentação são mais adequados para laboratórios que processam um tipo de amostra consistente em grande escala e priorizam a rapidez. Os métodos sem PCR são mais apropriados para laboratórios com acesso fiável a DNA de alta qualidade e alta quantidade e uma necessidade específica de redução de viés que eles proporcionam.
Comparação de Kits de Preparação de Biblioteca Disponíveis por Parâmetros Chave
Vários kits comercialmente disponíveis implementam cada uma das três estratégias acima. A escolha entre eles depende dos requisitos específicos do seu projeto.
| Tipo de Kit | Método | Intervalo de Entrada | Tempo Típico | Melhor Aplicação |
|---|---|---|---|---|
| PCR Livre (fragmentação padrão + ligadura) | Sonicação + ligadura, sem PCR | 100 ng – 1 µg | ~6 h | WGS onde o viés mínimo é crítico; DNA de alta qualidade |
| Baseado em PCR (fragmentação padrão + ligação) | Sonicação/enzimática + ligadura + PCR | 0,1 ng – 1 µg | ~4–6 h | Maior gama de aplicação; entrada flexível; FFPE com reparação |
| Baseada em tagmentação | Fragmentação de transposase + inserção de adaptador | 1–50 ng | ~3 h | Baixo input; fluxo de trabalho rápido; bactérias, vírus, genomas pequenos |
| Ultra-baixo-input / otimizado para cfDNA | Laço em haste ou ligadura especializada | 0,05–50 ng | ~2,5–3 h | cfDNA, biópsia líquida, célula única, entradas sub-nanogramas |
Conselhos práticos de seleçãoPara laboratórios que processam uma ampla gama de tipos de amostras, um kit de fragmentação + ligadura baseado em PCR com uma ampla faixa de entrada é a escolha mais versátil, cobrindo tudo, desde gDNA de alta qualidade até FFPE, cfDNA e bibliotecas de RNA-seq. Para laboratórios focados exclusivamente em WGS de DNA de alta qualidade, uma abordagem sem PCR oferece a melhor qualidade de dados com as taxas de duplicação mais baixas. Para laboratórios que priorizam a velocidade e a baixa entrada em tipos de amostras simples, a tagmentação é uma excelente opção que pode reduzir o tempo de manuseio em até 50%. Para projetos que envolvem cfDNA ou outras aplicações de entrada ultra-baixa, um kit especificamente otimizado para essas entradas deve ser a primeira consideração — usar um kit padrão baseado em PCR em cfDNA frequentemente leva à contaminação por dímeros de adaptadores, uma vez que os fragmentos de inserção curtos não proporcionam separação suficiente dos produtos de ligadura de adaptador-adaptador.
Considerações de custoO custo por biblioteca varia entre as três estratégias. Os kits de fragmentação baseados em PCR + kits de ligação geralmente têm o custo por reação mais alto, enquanto os kits de tagmentação costumam ser mais baixos. No entanto, o custo do kit por biblioteca deve ser ponderado em relação ao custo de uma corrida de sequenciação falhada. Uma pequena economia em reagentes de biblioteca é insignificante se produzir uma biblioteca que agrupa mal, gera uma baixa saída de dados ou introduz viés que compromete a interpretação biológica. O custo total do projeto—não o custo do reagente por biblioteca—é a métrica económica relevante.
Tipo de Amostra Especial 1 — FFPE e DNA Degradado
O DNA derivado de FFPE apresenta desafios únicos para a preparação de bibliotecas. A fixação em formalina causa ligações cruzadas que fragmentam o DNA e introduzem modificações de bases—mais notavelmente, a desaminação de citosina (C→T) que pode aparecer como mutações aparentes nos dados de sequenciação.
A preparação bem-sucedida de bibliotecas FFPE requer duas adaptações específicas:
- Reparação de danos antes da construção da bibliotecaUm passo de reparação pré-biblioteca utilizando uma mistura enzimática que contém uracilo-DNA glicosilase (UDG) e outras enzimas de reparação de danos remove citosinas desaminadas e repara quebras. Este passo é essencial para eliminar artefatos C→T que, de outra forma, apareceriam como variantes falso-positivas.
- Flexibilidade de entrada e redução do número de ciclosO DNA FFPE é tipicamente fragmentado para um tamanho médio de 200–400 bp, que já está dentro da faixa alvo para a maioria das bibliotecas Illumina. A ênfase deve estar em minimizar ciclos adicionais de amplificação para evitar viés adicional. Kits de fragmentação baseados em PCR + ligadura com módulos de reparo são a abordagem recomendada.
As bibliotecas FFPE normalmente apresentam uma distribuição de tamanhos de inserção mais ampla e menor complexidade do que as bibliotecas de DNA de alta qualidade. Um traço do Bioanalyzer que mostra um pico amplo de 150–600 bp, em vez de um pico agudo, é normal para amostras FFPE. O principal critério de controlo de qualidade não é a forma do pico, mas a presença de um pico de biblioteca claro acima de qualquer sinal de dímero de adaptador.
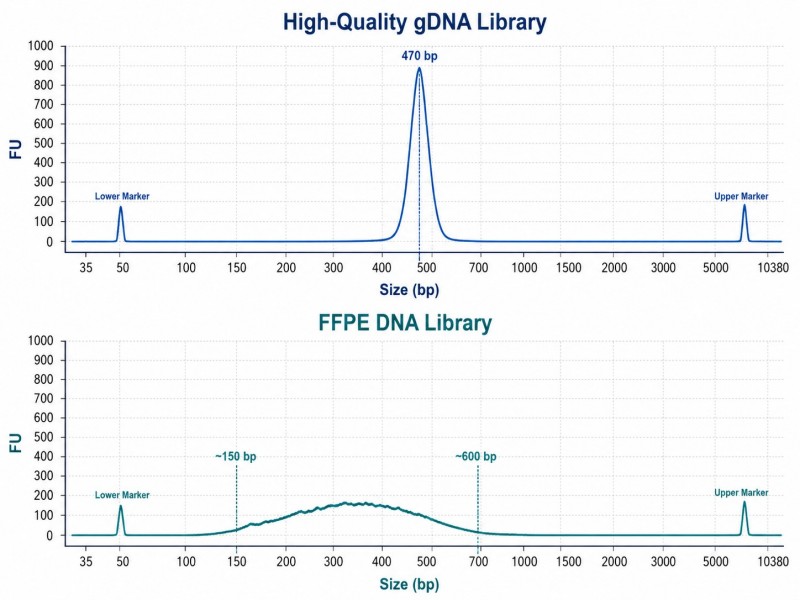 Figura 3. Comparação de traços do Bioanalyzer de DNA FFPE — DNA FFPE degradado versus traços de bibliotecas de DNA genómico de alta qualidade
Figura 3. Comparação de traços do Bioanalyzer de DNA FFPE — DNA FFPE degradado versus traços de bibliotecas de DNA genómico de alta qualidade
Legenda: Eletroforetograma comparativo do Bioanalyzer mostrando a forma de pico ampla típica de bibliotecas de DNA derivadas de FFPE em comparação com o pico agudo e bem definido de bibliotecas de DNA genómico de alta qualidade.
Tipo de Amostra Especial 2 — cfDNA e Amostras de Ultra Baixa Entrada
O DNA livre de células circulantes (cfDNA) está presente no plasma em concentrações muito baixas (tipicamente 1–50 ng por mL de sangue) e consiste em fragmentos com uma média de ~167 pb—o comprimento do DNA envolto em um único nucleossoma. Estas características exigem uma abordagem fundamentalmente diferente para a preparação de bibliotecas em comparação com o DNA genómico.
Considerações chave para a preparação de bibliotecas de cfDNA:
- Controlo do dimmer do adaptadorPorque os fragmentos de cfDNA são curtos (~167 bp + adaptadores = ~300 bp na biblioteca final), qualquer contaminação por dímeros de adaptadores (~120 bp) está dentro da mesma faixa de tamanho e não pode ser efetivamente removida por seleção de tamanho. Kits com adaptadores em forma de haste-laço ou bolha reduzem a formação de dímeros de forma mais eficaz do que adaptadores em Y padrão para aplicações de cfDNA.
- Otimização do ciclo de PCRA entrada de cfDNA é demasiado baixa para métodos sem PCR, pelo que a amplificação é necessária. No entanto, cada ciclo adicional de PCR corre o risco de introduzir viés e duplicados. O equilíbrio ideal é tipicamente de 8 a 12 ciclos, calibrado para produzir um rendimento de biblioteca suficiente sem exceder taxas de duplicação de 10 a 15%.
- Sensibilidade à mudança de índice: Porque cada molécula numa biblioteca de cfDNA é valiosa, a troca de índices que atribui erroneamente leituras entre amostras é particularmente prejudicial. Índices duplos únicos (UDI) são fortemente recomendados para projetos multiplexados de cfDNA.
O perfil da biblioteca de cfDNA num Bioanalyzer deve mostrar um pico a aproximadamente 280–330 bp (inserção de 167 bp + adaptador de 120–140 bp). Um segundo pico a 120–140 bp indica contaminação por dímeros de adaptador e sugere a necessidade de otimização do protocolo.
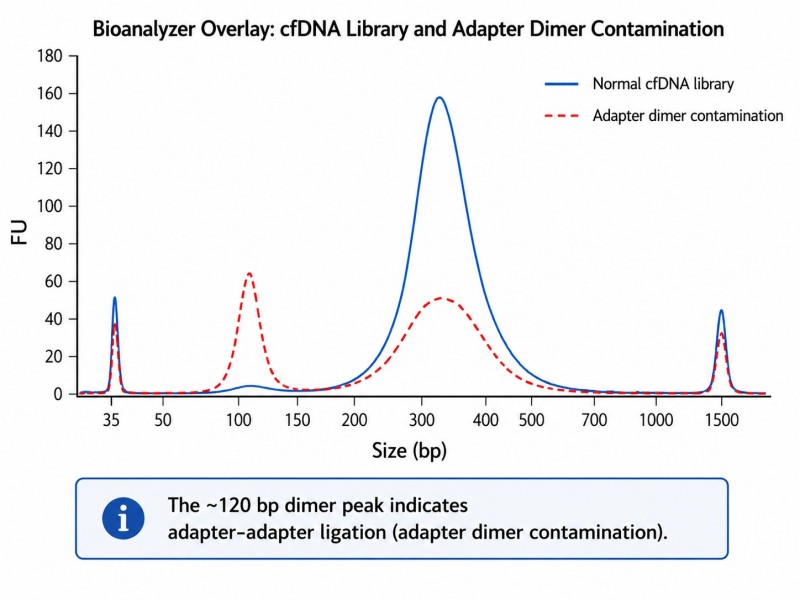 Figura 4. Traço do Bioanalyzer da biblioteca de cfDNA — pico típico da biblioteca e contaminação por dímeros de adaptadores
Figura 4. Traço do Bioanalyzer da biblioteca de cfDNA — pico típico da biblioteca e contaminação por dímeros de adaptadores
Legenda: Traço do bioanalisador de uma biblioteca de cfDNA mostrando o pico da biblioteca esperado em ~280–330 bp e um pico menor de dímero de adaptador em ~120–140 bp que indica contaminação, exigindo otimização do protocolo.
Comparação: cfDNA vs. preparação de biblioteca gDNA padrãoAs diferenças fundamentais entre a preparação de bibliotecas de cfDNA e gDNA vão além da quantidade de entrada. Como os fragmentos de cfDNA já são curtos e têm extremidades características (5-fosfato, grupos 3-hidroxilo resultantes da clivagem nucleossomal), a etapa de fragmentação é completamente omitida. O fluxo de trabalho de preparação da biblioteca começa diretamente com a reparação das extremidades e a adição de um "A", seguido pela ligação de adaptadores. A estratégia de limpeza também difere: as proporções das esferas SPRI devem ser cuidadosamente otimizadas para reter os curtos fragmentos de cfDNA enquanto remove os dímeros de adaptadores, que são apenas ~40–60 bp mais curtos do que os fragmentos da biblioteca alvo. Uma limpeza com esferas de dupla face—primeiro a 0,6–0,8× para remover fragmentos grandes, depois a 1,5–1,8× para capturar os fragmentos na faixa de cfDNA—é uma abordagem comum e eficaz.
WGS vs. Enriquecimento Direcionado vs. Amplicon — Como a Preparação de Bibliotecas Difere por Aplicação
O fluxo de trabalho de preparação da biblioteca muda dependendo do que vem a seguir.
| Aplicação | Objetivo de Preparação | Métrica Chave de QC | Requisitos de Entrada |
|---|---|---|---|
| Sequenciação do genoma completo | Cobertura uniforme com viés mínimo | Inserir distribuição de tamanhos, taxa de duplicação | 100 ng – 1 µg; preferencialmente sem PCR |
| Enriquecimento direcionado (WES, painéis) | Captura eficiente de regiões-alvo | % no alvo, uniformidade de cobertura | 10–200 ng |
| Sequenciação de amplicons | Amplificação equilibrada entre alvos | Ler o equilíbrio entre amplicões, dimerização de primers | 1–100 ng |
Para Preparação de biblioteca WGS, a prioridade é minimizar o viés experimental. Métodos sem PCR são preferidos quando a quantidade de entrada o permite. Os pontos de controlo críticos de QC são a distribuição do tamanho dos fragmentos (um pico estreito no tamanho alvo) e a taxa de duplicação (deve ser <10% para métodos sem PCR, <15% para métodos baseados em PCR).
Para preparação de biblioteca de enriquecimento direcionadoA biblioteca inicial é construída da mesma forma que uma biblioteca WGS, mas é adicionada uma etapa de captura por hibridização para captar regiões-alvo. A métrica crítica de controlo de qualidade muda de uniformidade de cobertura para eficiência de captura (% de leituras mapeadas para regiões-alvo). A preparação da biblioteca para enriquecimento direcionado deve produzir complexidade suficiente para evitar que leituras duplicadas dominem os dados em alvo após a captura.
Para preparação de biblioteca baseada em ampliconA biblioteca é gerada por PCR multiplex em vez de fragmentação e ligação de adaptadores. O principal desafio é equilibrar o desempenho dos primers em todos os alvos para evitar a perda de regiões específicas. O controlo de qualidade foca na uniformidade da cobertura em todo o conjunto de alvos — uma métrica padrão é a percentagem de amplicões dentro de 0,2×–2× da profundidade média de cobertura.
Uma nota sobre a preparação de bibliotecas de RNAEmbora o foco principal deste guia seja a preparação de bibliotecas de DNA, a preparação de bibliotecas de RNA-seq segue um fluxo de trabalho diferente. Em vez de fragmentação antes da ligação do adaptador, o RNA é primeiro convertido em cDNA através da transcrição reversa, e depois fragmentado (ou fragmentado antes da transcrição reversa, dependendo do protocolo). Os passos de ligação do adaptador e amplificação são semelhantes à preparação de bibliotecas de DNA, mas a especificidade da fita deve ser preservada se o desenho experimental exigir a distinção entre a fita de RNA original e a sua complementar.
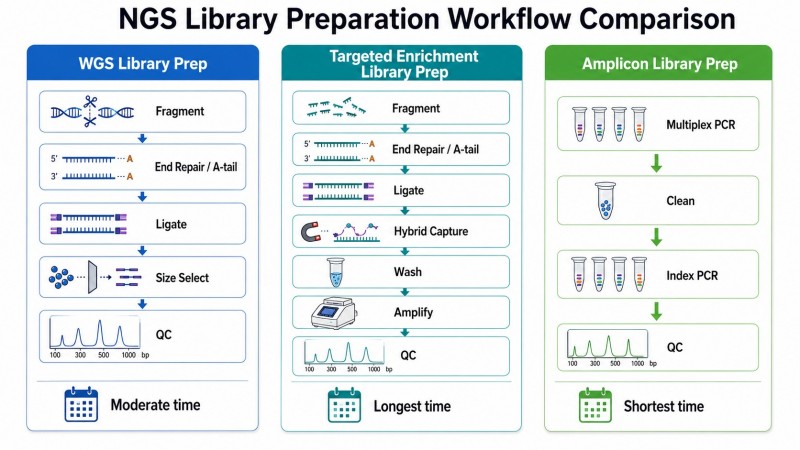 Figura 5. Preparação de biblioteca WGS vs preparação de biblioteca de enriquecimento direcionado vs preparação de biblioteca de amplicão — comparação de fluxos de trabalho
Figura 5. Preparação de biblioteca WGS vs preparação de biblioteca de enriquecimento direcionado vs preparação de biblioteca de amplicão — comparação de fluxos de trabalho
Diagrama de fluxo comparativo mostrando os três principais caminhos de preparação de bibliotecas NGS—WGS, enriquecimento direcionado e amplicon—com diferenças na estratégia de fragmentação, etapas de captura/amplificação e principais métricas de controlo de qualidade para cada um.
Vários kits de preparação de bibliotecas de RNA disponíveis comercialmente alcançam isso através de diferentes estratégias químicas. A QC da biblioteca de RNA-seq também deve avaliar a eficiência da depleção de RNA ribossómico, que é uma métrica de qualidade crítica não aplicável à preparação de bibliotecas de DNA.
Estratégia de multiplexação para projetos em loteAo preparar bibliotecas para um projeto de múltiplas amostras, a estratégia de codificação deve ser planeada antes do início da construção da biblioteca. Para projetos com 96 ou menos amostras por corrida de sequenciação, a indexação simples é geralmente suficiente, desde que as sequências de índice tenham diversidade adequada. Para projetos que excedem 96 amostras, ou quando as corridas multiplexadas serão combinadas para análise, recomenda-se fortemente o uso de índices duplos únicos (UDI). O UDI elimina o risco de "index hopping", que pode causar chamadas de variantes falso-positivas em designs de sequenciação agrupada. A escolha da estratégia de indexação afeta o fluxo de trabalho da preparação da biblioteca, uma vez que alguns conjuntos de índices requerem configurações específicas de adaptadores, que por sua vez afetam a química de ligadura e o protocolo de limpeza.
Resultados de QC da Biblioteca de Leitura — Um Guia Prático para Padrões de Falha Comuns
Aprender a interpretar os traços do Bioanalyzer ou do TapeStation é uma das habilidades mais úteis para quem trabalha com bibliotecas de NGS. Aqui estão os seis padrões de traço mais comuns e o que eles significam:
- Biblioteca normalUm pico único e simétrico na faixa de tamanho esperada. Para uma biblioteca de inserção padrão de 350 bp com adaptadores, isso significa um pico a aproximadamente 470 bp. Ombros menores de cada lado são aceitáveis.
- Pico do dímero do adaptadorUm segundo pico, menor, entre 120–140 bp (dependendo do design do adaptador). Isto indica que as moléculas do adaptador ligaram-se entre si durante o passo de ligadura, em vez de se ligarem ao DNA do inserto. Um pico de dímero <5% da massa total da biblioteca é geralmente aceitável; níveis mais altos requerem limpeza adicional ou revisão do protocolo.
- Pico deslocado para a esquerda (fragmentos curtos)O pico da biblioteca está abaixo da faixa de tamanho esperada. Isso geralmente indica uma sobre-fragmentação durante a etapa de cisalhamento ou tagmentação. Reduz o comprimento de leitura mapeável e pode exigir uma re-otimização das condições de fragmentação.
- Pico deslocado para a direita (fragmentos longos)O pico da biblioteca está acima da faixa de tamanho esperada. Isso indica subfragmentação ou seleção de tamanho ineficiente. Estas bibliotecas podem produzir uma densidade de clusters mais baixa na célula de fluxo porque fragmentos mais longos não se amplificam tão eficientemente.
- Pico largo (distribuição de tamanhos ampla)A sequência abrange mais de 500 bp sem um pico dominante. Isso é comum com DNA de entrada degradado, particularmente FFPE. É aceitável para bibliotecas FFPE, desde que a maioria dos fragmentos esteja dentro ou perto do comprimento da leitura de sequenciamento.
- Sem pico ou sinal muito baixoA biblioteca tem um rendimento extremamente baixo ou falhou completamente. As causas comuns incluem falha na ligadura do adaptador, DNA de entrada insuficiente ou enzima de ligadura degradada. Isto requer repetir a preparação da biblioteca com um controlo positivo para isolar a causa.
Exemplos de cada padrão de traço são mostrados abaixo.
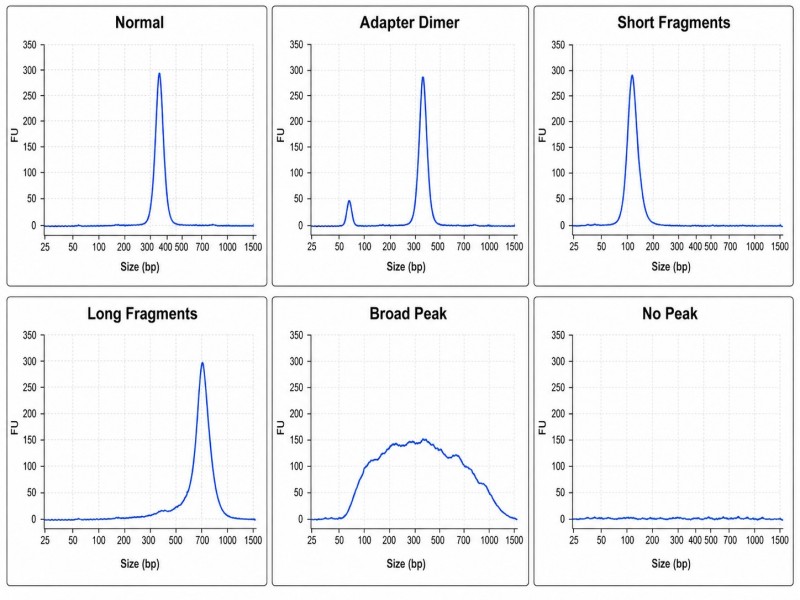 Figura 6. Traços de biblioteca do Bioanalyzer normais vs anormais — seis padrões de traço comuns com anotações
Figura 6. Traços de biblioteca do Bioanalyzer normais vs anormais — seis padrões de traço comuns com anotações
Legenda: Seis padrões de traço comuns do Bioanalyzer para bibliotecas de NGS: pico de biblioteca normal, contaminação por dímeros de adaptadores, deslocado para a esquerda (sobre-fragmentado), deslocado para a direita (sub-fragmentado), pico amplo (entrada degradada) e biblioteca falhada sem sinal detectável.
Como Otimizar os Rendimentos de Bibliotecas para Amostras de Baixo Input
Uma das questões práticas mais comuns que os investigadores enfrentam é como maximizar o rendimento da biblioteca quando o material de entrada é limitado. A recomendação padrão de "usar mais DNA de entrada" muitas vezes não é uma opção, por isso são necessárias estratégias alternativas.
Minimizar perdas em cada etapa de limpeza.Cada etapa de purificação com esferas SPRI perde 10–20% do material da biblioteca, mesmo em condições ótimas. Para amostras de baixo input, reduzir o número de etapas de purificação pode melhorar significativamente o rendimento final. Alguns protocolos combinam a purificação pós-ligação e pós-PCR em um único passo com esferas, à custa de uma distribuição de tamanhos ligeiramente mais ampla. Quando o input é criticamente limitado (sub-10 ng), esta compensação geralmente vale a pena aceitar.
Otimizar o número de ciclos de PCRA relação entre os ciclos de PCR e o rendimento da biblioteca não é linear. Os primeiros ciclos produzem uma amplificação exponencial; após 10–12 ciclos, ciclos adicionais acrescentam relativamente pouco rendimento enquanto aumentam significativamente as taxas de duplicação. Para entradas muito baixas (<5 ng), podem ser necessários 12–14 ciclos. Para entradas acima de 10 ng, 8–10 ciclos são tipicamente suficientes. Monitorizar a curva de amplificação por qPCR ou realizar um teste piloto em pequena escala a 8, 10, 12 e 14 ciclos para um novo tipo de amostra ajuda a identificar o equilíbrio ideal.
Utilize moléculas transportadorasAdicionar um transportador (por exemplo, poliacrilamida linear ou glicogénio) durante os passos de precipitação com etanol pode melhorar a recuperação de DNA em baixa concentração. Isto é particularmente útil para amostras de cfDNA onde as quantidades de entrada estão na faixa de 1–10 ng e cada nanograma conta.
Considere fluxos de trabalho de tubo único.Alguns métodos de preparação de bibliotecas recentemente desenvolvidos realizam fragmentação, reparação de extremidades, adição de A e ligação de adaptadores em um único tubo, sem etapas intermédias de limpeza. Esses fluxos de trabalho em tubo único podem melhorar a eficiência de conversão para amostras de baixo input em 30–50% em comparação com protocolos de múltiplas etapas, em detrimento de um controle de tamanho menos preciso.
Automação e Escalabilidade na Preparação de Bibliotecas
À medida que os projetos de sequenciação crescem em escala, a preparação manual de bibliotecas torna-se um gargalo. Um laboratório que processa 384 bibliotecas por semana não pode dar-se ao luxo de preparar cada biblioteca à mão, e os efeitos de lote de diferentes operadores podem introduzir variações técnicas indesejadas.
As plataformas de automação de manuseio de líquidos (como as da Beckman Coulter, Hamilton ou Agilent) podem processar 96 ou 384 amostras em paralelo com volumes de reagentes e tempos de incubação consistentes. As principais considerações para a adoção da preparação automatizada de bibliotecas incluem:
- Compatibilidade de protocoloNem todos os kits de preparação de bibliotecas comerciais têm protocolos de automação validados. Selecionar um kit com um script de automação estabelecido reduz significativamente o tempo de desenvolvimento.
- Restrições de volumeOs manipuladores de líquidos automatizados têm volumes mínimos de pipetagem (tipicamente 0,5–2 µL). Kits com volumes de reação menores podem exigir a mudança para versões de maior volume para compatibilidade com a automação.
- Manipulação de esferas magnéticasA limpeza automatizada baseada em esferas é o passo mais desafiador a otimizar. O tempo de assentamento das esferas, o engajamento do íman e a velocidade de remoção do sobrenadante afetam todos a reprodutibilidade.
- Integração de QCOs fluxos de trabalho automatizados mais eficientes incluem etapas de controlo de qualidade em linha, como a leitura automatizada de placas para quantificação, para identificar bibliotecas com falhas antes de seguirem para sequenciação.
Para equipas de investigação que estão a construir projetos de sequenciação em grande escala, serviços de NGS com capacidades de preparação de bibliotecas automatizadas pode fornecer a capacidade de processamento e a reprodutibilidade que os métodos manuais não conseguem igualar.
Um Fluxo de Trabalho de QC Prático Antes da Sequenciação
Antes de submeter um lote de bibliotecas a uma corrida de sequenciação, deve ser aplicado um fluxo de trabalho de QC padronizado a cada biblioteca. O objetivo é identificar bibliotecas que provavelmente falharão antes que desperdicem a capacidade do fluxo de células.
- Quantificação por qPCREste é o método mais preciso para determinar a concentração de moléculas de biblioteca amplificáveis. Um resultado de qPCR que difere da medição do Qubit por mais de 3 vezes frequentemente indica contaminação por dímeros de adaptadores ou falha na ligadura.
- Distribuição de tamanho por eletroforese capilarUm traço de Bioanalyzer ou TapeStation confirma que os fragmentos estão dentro da faixa de tamanho esperada e que os níveis de dímeros de adaptadores são aceitáveis.
- Cálculo da molaridadeA concentração da qPCR é convertida em molaridade utilizando o tamanho médio do fragmento obtido na análise do Bioanalyzer, que é usado para calcular o volume de carga.
- Normalização de poolPara corridas multiplexadas, as bibliotecas individuais são agrupadas em quantidades equimolares, com um excesso de 10-20% preparado.
- Titração pilotoPara novos tipos de bibliotecas, é altamente recomendável realizar um teste piloto com 2-3 concentrações de carga no tipo de célula de fluxo pretendido.
Este fluxo de trabalho de QC leva aproximadamente 2-3 horas para 96 bibliotecas e deve ser considerado uma parte rotineira de cada projeto de NGS.
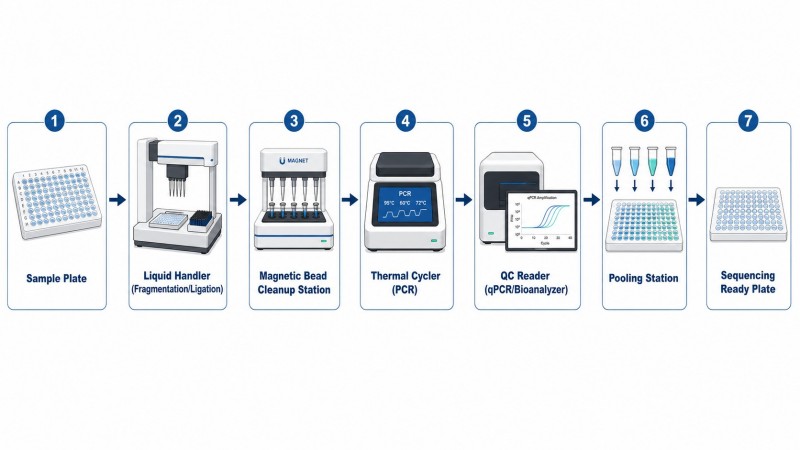 Figura 7. Fluxo de trabalho de automação para preparação de bibliotecas de alto rendimento
Figura 7. Fluxo de trabalho de automação para preparação de bibliotecas de alto rendimento
Legenda: Fluxo de trabalho automatizado de manuseio de líquidos para preparação de bibliotecas, mostrando os principais pontos de integração: compatibilidade de protocolos, restrições de volume, otimização do manuseio de esferas magnéticas e controlo de qualidade em linha para produção escalável.
Como a CD Genomics Apoia a Preparação de Bibliotecas
A CD Genomics oferece serviços abrangentes de preparação de bibliotecas que cobrem toda a gama de métodos e tipos de amostras compatíveis com Illumina.
Métodos disponíveisO nosso laboratório executa métodos de preparação de bibliotecas baseados em fragmentação + ligação por PCR, sem PCR e baseados em tagmentação, utilizando os kits comerciais mais amplamente adotados. A escolha do método é determinada pelo tipo de amostra e pelos requisitos do projeto.
Especialização em amostras especiaisAcumulámos uma vasta experiência em reparação de DNA FFPE e construção de bibliotecas, preparação de bibliotecas de cfDNA e de ultra-baixo input, e fluxos de trabalho otimizados para automação em projetos de grande escala. Os nossos protocolos estão validados para sangue, tecido, FFPE, cfDNA, células únicas, bem como amostras de plantas e microbianas.
QC da BibliotecaCada biblioteca passa por um rigoroso controlo de qualidade, incluindo análise de traços com Bioanalyzer ou TapeStation, quantificação por qPCR e confirmação da distribuição de tamanhos antes de prosseguir para a sequenciação.
EscalabilidadePara projetos de grandes lotes, implementamos fluxos de trabalho automatizados de manuseio de líquidos que garantem uma qualidade de biblioteca consistente em todas as amostras. Isso é particularmente valioso para projetos de múltiplos lotes, onde a reprodutibilidade de lote para lote é essencial.
Para mais detalhes, explore o nosso serviços de NGS ou contacte a nossa equipa para recomendações específicas do projeto.
Perguntas Frequentes
Qual é a diferença entre a preparação de bibliotecas baseada em PCR e a preparação de bibliotecas sem PCR?
As bibliotecas baseadas em PCR incluem uma etapa de amplificação que aumenta o rendimento, mas pode introduzir viés em regiões ricas em GC e aumentar as taxas de duplicação. As bibliotecas sem PCR dispensam a amplificação, produzindo uma cobertura mais uniforme, mas requerem uma maior quantidade de DNA de entrada (>100 ng).
Qual o melhor método de preparação de biblioteca para amostras de ADN FFPE?
A fragmentação baseada em PCR + ligação com um passo de reparação de DNA pré-biblioteca é recomendada para amostras FFPE. O passo de reparação remove artefatos de desaminação de citosina que, de outra forma, apareceriam como mutações falsas positivas.
A preparação de bibliotecas sem PCR pode ser utilizada com DNA de baixo input?
Não tipicamente. Métodos sem PCR requerem 100 ng a 1 µg de DNA de entrada. Para entradas abaixo de 100 ng, é necessário um método baseado em PCR para gerar um rendimento de biblioteca suficiente.
Como é que um dímero de adaptador aparece num traço de Bioanalyzer?
Um dímero de adaptador aparece como um pequeno pico a aproximadamente 120–140 pb, bem abaixo do pico esperado da biblioteca. Se o dímero constituir mais de 5% da massa total da biblioteca, recomenda-se uma limpeza adicional.
Como escolho entre tagmentação e fragmentação + ligadura?
A tagmentação oferece um fluxo de trabalho mais rápido e requisitos de entrada mais baixos, mas pode introduzir viés de GC. A fragmentação + ligadura proporciona uma cobertura mais uniforme em todo o conteúdo de GC e é a escolha preferida para WGS e aplicações que requerem uma representação genómica uniforme.
Qual é a quantidade mínima de DNA de entrada para a preparação padrão de bibliotecas?
Os kits padrão baseados em PCR normalmente requerem um input mínimo de 0,1 a 1 ng. Os kits de ultra-baixo input podem funcionar com apenas 50 pg. Os kits sem PCR requerem de 100 ng a 1 µg.
Como posso remover dimers de adaptadores da minha biblioteca?
Os dímeros de adaptadores podem ser reduzidos otimizando a proporção de limpeza com esferas SPRI (proporções mais altas retêm mais dímeros), utilizando uma seleção de tamanho de dois lados ou adicionando um passo de seleção de tamanho baseado em gel para casos teimosos.
Quais métricas de QC devo reportar para a preparação da biblioteca de WGS?
No mínimo: concentração da biblioteca (a partir de qPCR), distribuição do tamanho dos fragmentos (a partir do Bioanalyzer) e taxa de duplicação (a partir de uma corrida de QC de sequenciamento raso ou estimativa computacional).
A CD Genomics pode preparar bibliotecas a partir de cfDNA?
Sim. Oferecemos preparação de bibliotecas otimizada para cfDNA utilizando protocolos de entrada ultra-baixa e designs de adaptadores em forma de laço que minimizam a formação de dímeros de adaptador em bibliotecas de fragmentos curtos.
Quanto tempo demora uma preparação típica de biblioteca?
Um protocolo padrão de fragmentação + ligação baseado em PCR leva de 4 a 6 horas desde a entrada de DNA até a biblioteca pronta para sequenciamento. Métodos de tagmentação podem ser concluídos em aproximadamente 3 horas. Métodos sem PCR requerem de 5 a 7 horas devido a etapas adicionais de limpeza necessárias para remover adaptadores não incorporados, sem a opção de limpeza baseada em PCR.
Qual é a diferença entre a preparação de biblioteca de índice único e de índice duplo?
As bibliotecas de índice único utilizam uma sequência de código de barras por amostra, enquanto as bibliotecas de índice duplo utilizam duas sequências de código de barras independentes (uma em cada adaptador). O índice duplo reduz o risco de atribuição incorreta de amostras quando várias bibliotecas são agrupadas para sequenciação, particularmente em plataformas de células de fluxo padronizadas, onde a troca de índices é mais comum.
Quais são as causas das altas taxas de duplicação nos dados de sequenciação?
Taxas de duplicação elevadas (>20%) são mais frequentemente causadas por DNA de entrada insuficiente, ciclos de PCR excessivos ou baixa complexidade da biblioteca inicial. Para bibliotecas de WGS, métodos sem PCR produzem as taxas de duplicação mais baixas, enquanto métodos baseados em PCR com entradas abaixo da faixa recomendada são os mais propensos a este problema.
Apenas para uso em investigação.
Referências:
- Melhores práticas para a preparação de bibliotecas Illumina. Protocolos Actuais em Genética Humana. 2019;102(1):e86.
- Comparação do viés de sequenciação dos kits de preparação de bibliotecas atualmente disponíveis. Pesquisa de DNA. 2019;26(5):391-402.
- Análise comparativa de abordagens de preparação de bibliotecas para DNA de FFPE. Relatórios Científicos. 2025;15:12992.
- Uma análise comparativa das abordagens de preparação de bibliotecas para amostras de baixo input. BMC Genómica. 2018;19:763.


