
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
O que é CITE-Seq?
O CITE-seq (indexação celular de transcriptomas e epítopos) representa um avanço na sequenciação de células únicas, permitindo a captura simultânea de transcritos e uma porção de proteínas de superfície da membrana. Este método inovador complementa a sequenciação de RNA de células únicas da 10X Genomics ao incorporar a detecção de proteínas de superfície celular. Análises estatísticas revelam uma forte correlação entre a categorização celular baseada em RNA e ADT, indicando perda mínima de sinal na detecção de ADT, facilitando assim a identificação precisa da classe celular. Particularmente vantajoso para análises que envolvem um alto número de células propensas a ambiguidade, o CITE-seq melhora a capacidade de distinguir entre tipos celulares sem comprometer a integridade do sinal.
Simultaneamente, a tecnologia de marcação de hashing celular permite a realização de mistura de múltiplas amostras para a detecção de CITE-seq, seguida pela separação dos dados de células únicas. Isso facilita a subsequente identificação e caracterização de linhagens celulares e proteínas de superfície capturadas por anticorpos, aproveitando as sequências de tags HTO ou ADT, respetivamente.
Vantagens do CITE-Seq
- Versatilidade na Detecção de Proteínas: O CITE-seq possui a capacidade de detectar mais de 100 proteínas de superfície simultaneamente, integrando-se perfeitamente com plataformas comerciais. Isso supera as limitações da separação por fluxo, oferecendo uma gama ilimitada de anticorpos.
- Melhoria na Tipificação Celular: O CITE-seq permite uma identificação mais clara dos tipos celulares, facilita a anotação de tipos celulares raros e promove a descoberta de novas subpopulações celulares.
- Mitigação de Dropout: A integração com a tecnologia de rotulagem de Hashing celular aborda efetivamente a questão do dropout, melhorando a qualidade dos dados.
- Integração de mRNA e Proteína: O CITE-seq alcança a detecção conjunta de multi-ômicas dentro da mesma célula, fornecendo informações simultâneas de mRNA e proteínas de superfície celular. Esta abordagem abrangente supera a sequenciação de RNA de células únicas tradicional em termos de informação.
Além disso, a 10X Genomics oferece um sistema de rotulagem acoplado a ADTs bem estabelecido e um quadro procedimental detalhado para o CITE-seq. Aproveitando a tecnologia de Código de Características, realiza eficientemente estudos de transcriptoma e proteínas de superfície em células imunes. Adicionalmente, produtos compatíveis, como anticorpos acoplados a oligonucleotídeos e MHC acoplados a oligonucleotídeos, facilitam ainda mais a análise multi-ômica de células únicas.
Aplicações do CITE-Seq
O CITE-seq serve como uma ferramenta poderosa para integrar informações de proteínas com genes, auxiliando na caracterização precisa dos estados celulares.
O método de análise de Vizinhos Mais Próximos Ponderados (WNN) representa uma abordagem sofisticada para harmonizar dados de multi-ômicas dentro de células individuais, resultando em uma definição abrangente do estado celular. Este método opera de forma não supervisionada, inicialmente computando pesos histológicos específicos de células através de predição cruzada em múltiplas camadas ômicas. Esses pesos, que encapsulam a importância relativa de cada componente de informação histológica, informam subsequentemente a métrica de distância entre células, resultando na construção de um gráfico de vizinhança WNN—denominado gráfico WNN—que retrata a paisagem multi-ômica dentro de células únicas. O gráfico WNN encontra utilidade em várias tarefas de análise subsequente na análise de dados de células únicas, incluindo visualização UMAP, análise de agrupamento e construção de séries temporais citomiméticas.
- Determinação da Identidade Celular
Inicialmente, os dados de RNA foram processados individualmente, e os dados de RNA+ADT foram analisados em conjunto utilizando a abordagem de Vizinhos Mais Próximos Ponderados (WNN). A anotação utilizando marcadores clássicos revelou que a incorporação de dados de ADT durante a análise levou a uma diferenciação e anotação melhoradas dos tipos celulares. Notavelmente, a introdução da conversão de ADT destacou diferenças mais distintas entre subpopulações de células T em comparação com RNA sozinho. A combinação de gráficos de violino de pesos de RNA e ADT dentro de cada tipo celular indicou pesos de ADT elevados dentro de Macrófagos (MPs) e subpopulações de células T. A integração de ADT para redução de dimensionalidade demonstrou uma discriminação melhorada entre subpopulações em comparação com RNA sozinho, oferecendo validação extensiva de proteínas e identificação de marcadores de superfície críticos para cada subpopulação. Estudos anteriores corroboraram que o CITE-seq melhora a resolução populacional, distinguindo efetivamente subpopulações semelhantes e até mesmo revelando novas populações de macrófagos PD-L1/PD-L2+ correlacionadas com resultados clínicos.
- Delineação de Subpopulações
Subsequentemente, a delineação de subpopulações e a validação das descobertas do scRNA-seq foram realizadas. Dado os pesos de proteínas mais altos observados em células T, buscou-se uma subdivisão adicional das subpopulações de células T. A validação das subpopulações identificadas por scRNA através dos resultados do CITE-seq revelou uma separação mais pronunciada das subpopulações em gráficos UMAP com base nos dados de proteínas. Gráficos de violino de pesos de RNA e ADT destacaram o papel da expressão de proteínas na distinção de subpopulações durante a subdivisão de células T. Além disso, a validação extensiva de proteínas facilitou a identificação de marcadores de superfície chave para cada subpopulação.
- Exploração de Mecanismos através de Análise Funcional
A subdivisão adicional de subgrupos de NK revelou que a incorporação de características de ADT facilitou uma melhor discriminação de subgrupos, com expressão distinta de marcadores de proteínas observada entre subgrupos. Por exemplo, o cluster1 exibiu expressão específica do marcador de proteína CD8a, enquanto o cluster5 mostrou um enriquecimento significativo na via de toxicidade mediada por células NK. Notavelmente, não houve diferenças significativas na expressão do gene CD8A entre os clusters.
Estudo de Caso: Análise de Células Únicas do Fígado
Em um esforço inovador, a equipe de pesquisa uniu o CITE-seq de células únicas com tecnologias complementares para mapear o proteoma espacial de células únicas do fígado humano e de camundongos, lançando luz sobre a população de macrófagos associados a lipídios. Aprofundando-se na expressão de mRNA e proteínas em granularidade de células únicas, 18 amostras passaram por CITE-seq e subsequente análise de dados, culminando na identificação eficaz de 17 tipos celulares distintos. Simultaneamente, o sistema de reconhecimento de anticorpos ADT do CITE-seq capturou habilmente células de Kupffer, e através de uma análise meticulosa da população celular, a proteína VSIG4 emergiu como o principal marcador para células de Kupffer humanas dentro do conjunto de dados do CITE-seq.
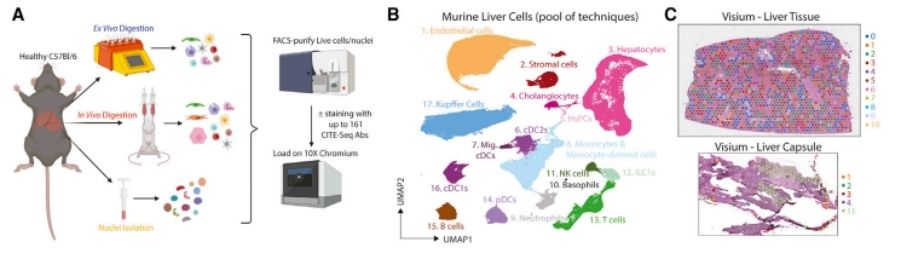 Um atlas proteogenômico do fígado murino saudável. (Guilliams et al., 2022)
Um atlas proteogenômico do fígado murino saudável. (Guilliams et al., 2022)
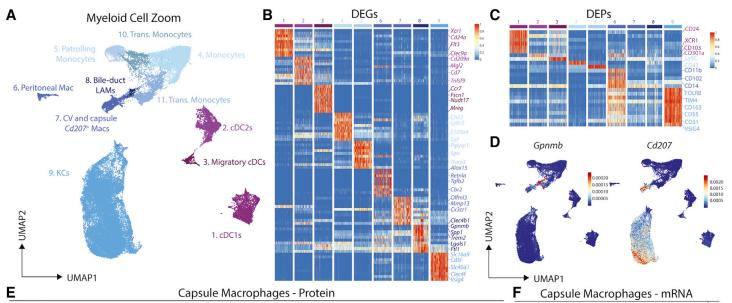 Uma população de macrófagos reside ao redor do ducto biliar no fígado murino saudável. (Guilliams et al., 2022)
Uma população de macrófagos reside ao redor do ducto biliar no fígado murino saudável. (Guilliams et al., 2022)
Estudo de Caso: Investigação de Células Únicas do Câncer de Pulmão
Este estudo apresenta uma análise abrangente da maior coorte de câncer de pulmão não pequenas células (NSCLC) até à data, empregando metodologias de sequenciação de RNA de células únicas (scRNA-seq) e CITE-seq. Ao aprofundar-se nas respostas imunes compartilhadas entre pacientes, a pesquisa identificou fatores comuns enquanto descobria efeitos moduladores imunes distintos da carga de mutação tumoral e das mutações TP53. Consequentemente, modelos refinados foram elaborados para prever respostas à imunoterapia com maior precisão.
A paisagem transcricional do microambiente tumoral foi meticulosamente explorada através de análises de scRNA-seq e CITE-seq, revelando um espectro de estados transcricionais.
Especificamente, os dados do CITE-seq destacaram características únicas das células T CD8+, assemelhando-se a células NK, delineando assim subtipos de células T dentro de tecidos normais e cancerígenos. Notavelmente, os tecidos cancerígenos exibiram uma gama mais ampla de subtipos de células T, abrangendo células T ativadoras, em ciclo e reguladoras.
Aproveitando as percepções dos dados do CITE-seq e outras investigações multi-ômicas de células únicas, um mapa de resposta imune de células únicas foi construído para o câncer de pulmão. Este mapa delineou uma assinatura de ativação imune, apontando a interação entre cargas antigênicas associadas ao tumor e estados de mutação condutora. Além disso, um modelo foi desenvolvido utilizando o eixo LCAM, oferecendo uma medida mais direta da especificidade antigênica e ativação imune anti-tumoral. Este modelo refinado promete uma precisão aprimorada na definição da ativação imune dentro de tumores específicos de antígenos.
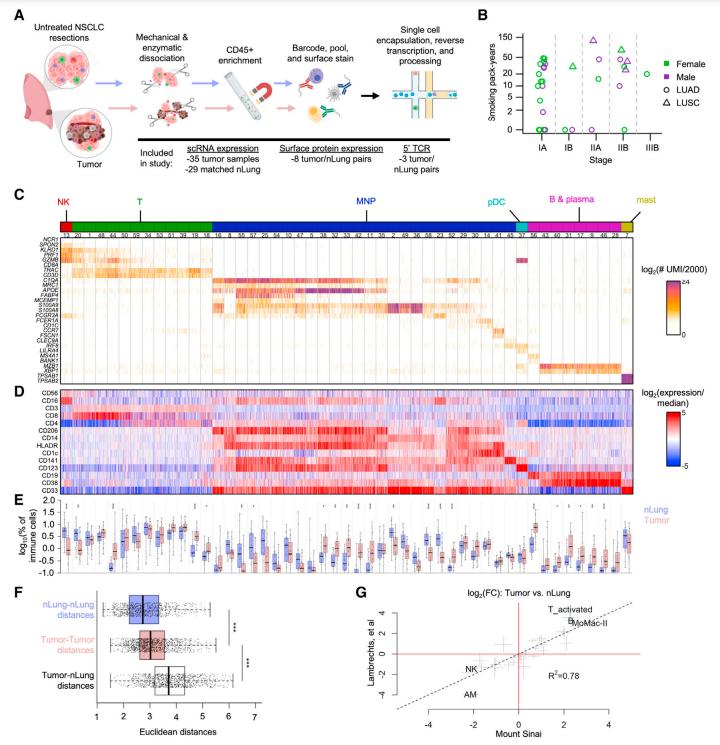 scRNA-seq e CITE-seq estabelecem a diversidade de estados transcricionais no microambiente tumoral. (Leader et al., 2021)
scRNA-seq e CITE-seq estabelecem a diversidade de estados transcricionais no microambiente tumoral. (Leader et al., 2021)
Referências:
- Guilliams, Martin, et al. "A proteogenômica espacial revela nichos hepáticos distintos e evolutivamente conservados de macrófagos." Cell 185.2 (2022): 379-396.
- Leader, Andrew M., et al. "Análise de células únicas de lesões humanas de câncer de pulmão não pequenas células refina a classificação tumoral e a estratificação de pacientes." Cancer cell 39.12 (2021): 1594-1609.


