
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
Construção de Bibliotecas para Sequenciação de Nova Geração (NGS)
A construção da biblioteca é o passo mais suscetível a variabilidade no fluxo de trabalho de NGS e a principal causa de falhas em corridas de sequenciação. Uma biblioteca bem construída com parâmetros otimizados—método de fragmentação, relação molar de adaptadores, protocolo de seleção de tamanho e número de ciclos de amplificação—pode ser a diferença entre um projeto que fornece dados prontos para publicação na primeira corrida e um que requer resolução de problemas repetida e re-sequenciação.
O custo da construção subótima de bibliotecas vai além da despesa direta com reagentes. Uma biblioteca com um conteúdo excessivo de dímeros de adaptadores desperdiça leituras de sequenciamento; uma com muitos duplicados de PCR reduz a profundidade de cobertura efetiva; uma biblioteca com seleção de tamanho incorreta produz taxas de mapeamento mais baixas. Quando essas ineficiências se acumulam em um projeto com múltiplas amostras, a perda de dados cumulativa pode ser substancial—equivalente a perder faixas inteiras ou células de fluxo de capacidade de sequenciamento. Para um projeto típico de WGS com 96 amostras a 30× de cobertura, uma perda de 20% em leituras utilizáveis devido a problemas de qualidade da biblioteca traduz-se em aproximadamente 0,5-1 Tb de dados de sequenciamento desperdiçados e dezenas de milhares de dólares em custos de sequenciamento irrecuperáveis.
Este guia é escrito para investigadores que já compreendem os passos básicos da construção de bibliotecas, mas que necessitam de orientação quantitativa sobre a otimização de parâmetros. Abrange os principais parâmetros ajustáveis em cada etapa do fluxo de trabalho, os compromissos associados a cada escolha e as métricas de controlo de qualidade que distinguem uma boa biblioteca de uma falhada.
O foco é deliberadamente prático: após ler este guia, deverá ser capaz de justificar a sua escolha de método de fragmentação, calcular uma relação molar adequada entre adaptador e inserto, selecionar a proporção certa de esferas SPRI para o tamanho do fragmento alvo e interpretar um traço do Bioanalyzer para determinar se a sua biblioteca passa no controlo de qualidade.
Os parâmetros discutidos neste guia são pontos de partida—não regras fixas. Cada laboratório, tipo de amostra e plataforma de sequenciação pode exigir pequenos ajustes a estes valores recomendados. A abordagem mais eficaz é estabelecer uma linha de base utilizando os parâmetros recomendados, e depois titrar variáveis-chave (ciclos de PCR, proporções de esferas) através de pequenos experimentos piloto para determinar as configurações ótimas para o seu fluxo de trabalho e tipo de amostra específicos.
O Fluxo de Trabalho Principal em Resumo
Todos os fluxos de trabalho de construção de bibliotecas da Illumina seguem a mesma sequência de etapas, independentemente do kit ou protocolo específico ou do tipo de amostra inicial:
- FragmentaçãoO DNA é partido em fragmentos de tamanho alvo.
- Reparação de extremidades e A-tailingAs extremidades dos fragmentos são embotadas, fosforiladas e possuem uma cauda de adenina.
- Ligação de adaptadoresOs adaptadores de sequenciação são ligados aos fragmentos.
- Limpeza e seleção de tamanhosAdaptadores e fragmentos não incorporados fora do intervalo alvo são removidos usando esferas magnéticas SPRI.
- Amplificação de bibliotecaA amplificação por PCR aumenta o rendimento da biblioteca (métodos sem PCR saltam esta etapa).
- QC da BibliotecaA biblioteca final é quantificada e verificada quanto à qualidade.
Os passos principais do fluxo de trabalho de construção de bibliotecas da Illumina—fragmentação, reparação das extremidades, ligação de adaptadores, limpeza, amplificação e controlo de qualidade—podem ser visualizados como um processo linear. O DNA de entrada entra à esquerda, passa por cada etapa de transformação e sai à direita como uma biblioteca pronta para sequenciar. A qualidade da biblioteca final depende do efeito cumulativo das escolhas de parâmetros em cada etapa, sendo que os passos iniciais têm o maior impacto, uma vez que os erros se propagam para a frente através do fluxo de trabalho.
Cada um destes passos tem parâmetros ajustáveis que afetam significativamente a qualidade final da biblioteca. As seções seguintes fornecem orientações quantitativas para cada escolha de parâmetro.
 Figura 1. Fluxo de trabalho de construção de biblioteca Illumina em seis etapas — desde a fragmentação do DNA até o controlo de qualidade final.
Figura 1. Fluxo de trabalho de construção de biblioteca Illumina em seis etapas — desde a fragmentação do DNA até o controlo de qualidade final.
Legenda: O fluxo de trabalho de construção de bibliotecas Illumina em seis etapas, mostrando a progressão linear desde a fragmentação até a reparação das extremidades, ligação de adaptadores, seleção de tamanho, amplificação e controlo de qualidade final, com erros em estágios iniciais a propagar-se ao longo do processo.
Fragmentação — Comparação de Abordagens Mecânicas, Enzimáticas e de Tagmentação
A fragmentação é o primeiro parâmetro variável no fluxo de trabalho de construção de bibliotecas e um que tem um impacto mensurável na qualidade dos dados subsequentes. Estão disponíveis três abordagens, cada uma com características distintas que afetam a uniformidade da cobertura, a reprodutibilidade e a flexibilidade de entrada.
Fragmentação mecânica utiliza energia acústica (Covaris) ou nebulização para fragmentar o DNA em fragmentos. O processo é puramente físico—nenhuma enzima está envolvida—o que significa que produz essencialmente nenhum viés dependente da sequência. Isso torna a fragmentação mecânica o método preferido para projetos de WGS onde uma cobertura uniforme em todas as regiões genómicas, incluindo promotores de alto GC e intrões de baixo GC, é essencial. As desvantagens são o custo mais elevado do instrumento, o tempo de processamento mais longo e a menor capacidade em comparação com métodos enzimáticos.
Fragmentação enzimática utiliza um cocktail de nucleases (tipicamente NEBNext Ultra II ou KAPA HyperPlus) para introduzir quebras de dupla fita. A fragmentação baseada em enzimas é mais escalável do que a fragmentação mecânica e funciona numa gama de entradas mais ampla, desde quantidades sub-nanogramas até vários microgramas. O perfil de viés depende da formulação da enzima — algumas misturas de enzimas produzem qualidade quase mecânica com um rendimento significativamente mais elevado.
Tagmentação (baseada em Tn5) usa uma transposase Tn5 hiperativa que fragmenta simultaneamente o DNA e insere sequências de adaptadores numa única reação. Isto reduz o tempo de manuseio de aproximadamente 30 minutos para menos de 5 minutos e funciona com apenas 1 ng de DNA de entrada. O compromisso é que o Tn5 apresenta um viés mensurável em regiões de baixo GC, onde a cobertura pode cair para 60-70% da média do genoma. A tagmentação é mais adequada para aplicações onde a velocidade e a baixa entrada são priorizadas em relação à uniformidade absoluta da cobertura.
| Parâmetro | Mecânico (Covaris) | Enzimático | Tagmentação (Tn5) |
|---|---|---|---|
| Intervalo de ADN de entrada | 50 ng – 5 µg | 0,5 ng – 1 µg | 1–50 ng |
| viés de GC | Mais baixo | Baixo a moderado | Moderado (regiões de baixo GC) |
| Reproduzibilidade (CV) | <10% | 10–20% | 15–25% |
| Tempo prático | 10–15 min (mais instrumento) | 5–10 min | 2–5 min |
| Mais adequado para | WGS, amostras de alto valor | Uso geral, FFPE, cfDNA | Baixo input, triagem rápida, genomas bacterianos |
Lógica de seleçãoPara projetos de WGS onde a cobertura uniforme em relação ao conteúdo de GC é crítica, a fragmentação mecânica produz o menor viés. Para fluxos de trabalho de uso geral que processam tipos de amostras diversos, a fragmentação enzimática oferece o melhor equilíbrio entre conveniência e qualidade. A tagmentação é ideal para aplicações de baixo input ou de alto rendimento onde a velocidade é priorizada em relação à uniformidade da cobertura.
Ao selecionar um método de fragmentação, a aplicação subsequente dita a escolha apropriada. Por exemplo, Projetos de sequenciação de RNA utilize a fragmentação de cDNA induzida por calor em vez de qualquer um dos três métodos de fragmentação de DNA, tornando o passo de fragmentação fundamentalmente diferente dos fluxos de trabalho de bibliotecas de DNA.
Verificação da qualidade da fragmentaçãoAntes de prosseguir com a reparação e ligadura, é aconselhável verificar a distribuição do tamanho dos fragmentos utilizando um Bioanalisador ou TapeStation. O traço deve mostrar uma distribuição unimodal centrada no tamanho alvo. Uma distribuição bimodal ou um pico amplo que abranja >500 bp indica condições de fragmentação subótimas e pode exigir o ajuste do tempo de cisalhamento ou da concentração da enzima antes de prosseguir com o restante do fluxo de trabalho.
Os investigadores que planeiam projetos de grande escala podem aproveitar serviços de sequenciação do genoma completo onde a fragmentação e a construção da biblioteca são otimizadas para cada tipo de amostra.
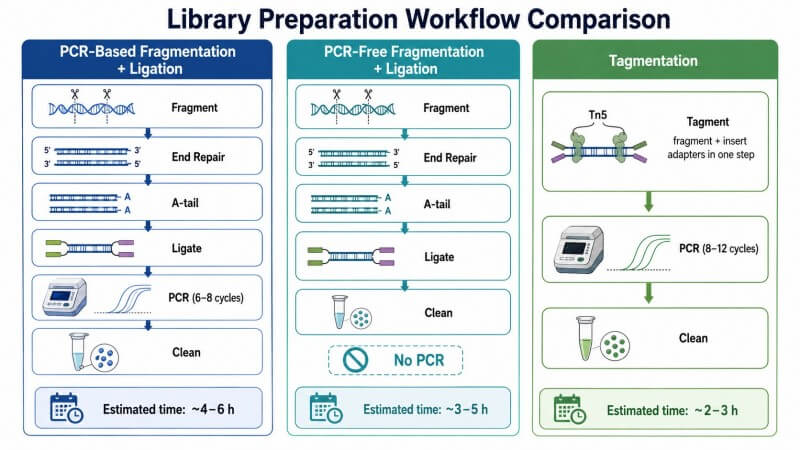 Figura 2. Comparação dos métodos de fragmentação — perfis de viés de cobertura em função do conteúdo de GC para abordagens mecânicas, enzimáticas e de tagmentação.
Figura 2. Comparação dos métodos de fragmentação — perfis de viés de cobertura em função do conteúdo de GC para abordagens mecânicas, enzimáticas e de tagmentação.
Legenda: Perfis de viés de cobertura comparativa em função do conteúdo de GC para três métodos de fragmentação, mostrando que a fragmentação mecânica produz a cobertura mais uniforme, a fragmentação enzimática apresenta um viés baixo a moderado, e a tagmentação apresenta um viés mensurável em regiões de baixo GC.
Ligação de Adaptadores — Otimização da Proporção Molar
A relação molar entre adaptadores e fragmentos de DNA de inserção é um dos parâmetros mais frequentemente mal otimizados na construção de bibliotecas. Se estiver errada, pode-se produzir dímeros de adaptadores (demasiado adaptador) ou bibliotecas etiquetadas de forma ineficiente (demasiado pouco adaptador). A relação ideal depende da concentração das extremidades de fragmentos ligáveis, que é determinada tanto pela massa de DNA de entrada quanto pelo tamanho médio dos fragmentos. Uma dada massa de fragmentos curtos tem mais extremidades por unidade de massa do que a mesma massa de fragmentos longos, por isso a relação molar deve levar em conta o número de extremidades disponíveis.
Para a construção de bibliotecas padrão da Illumina, a relação molar recomendada entre adaptador e inserto varia entre 10:1 e 50:1, dependendo da quantidade de entrada:
- >500 ng de entrada → proporção de 10:1 a 20:1 (eficiência de ligação suficiente, risco mínimo de dímeros)
- 100–500 ng de entrada → proporção de 20:1 a 30:1
- 10–100 ng de entrada → razão de 30:1 a 50:1 (razão mais alta necessária para compensar a menor probabilidade de colisão)
- <10 ng de entrada → proporção de 50:1 a 100:1; o dímero do adaptador torna-se um risco significativo, e kits especializados de baixa entrada com adaptadores em stem-loop são recomendados em vez de adaptadores Y padrão.
Adaptadores em Y vs. adaptadores em laço de cauleOs adaptadores Y padrão têm dois braços não complementares que criam uma estrutura em forma de garfo. Durante a ligadura, ambos os braços podem participar em eventos de ligadura, e quando não há inserto presente, os dois braços podem ligar-se entre si para formar dímeros de adaptador. Os adaptadores em laço de haste utilizam uma estrutura de alça que impede a ligadura de adaptador a adaptador, reduzindo a formação de dímeros em 50-80% em comparação com os adaptadores Y. Para a construção de bibliotecas de baixo input, onde os dímeros de adaptador são o principal modo de falha, os adaptadores em laço de haste proporcionam uma melhoria significativa na qualidade.
A contaminação por dímeros de adaptadores é detectável num traço do Bioanalyzer como um pico entre 120–140 bp. A níveis abaixo de 5% da massa total da biblioteca, os dímeros têm um impacto mínimo na sequenciação; acima de 5%, consomem leituras de sequenciação sem produzir dados alinháveis, reduzindo efetivamente a saída de dados utilizáveis do projeto. A níveis acima de 20%, os dímeros de adaptadores tornam-se a espécie dominante na biblioteca, e a corrida de sequenciação produzirá principalmente leituras não alinháveis.
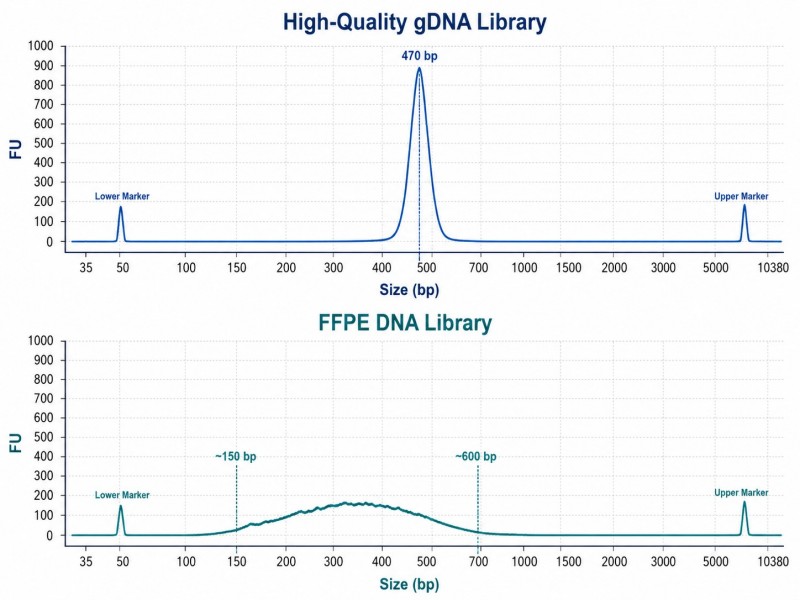 Figura 3. Rácio molar do adaptador vs. rendimento da biblioteca e formação de dímeros — relação quantitativa
Figura 3. Rácio molar do adaptador vs. rendimento da biblioteca e formação de dímeros — relação quantitativa
Relação quantitativa entre a razão molar de adaptador para inserto e o rendimento da biblioteca (azul) e a formação de dímeros de adaptador (vermelho), mostrando a janela de operação ideal entre 10:1 e 50:1, dependendo da quantidade de DNA de entrada.
Reparo de Extremidades e A-Tailing — Um Passo Intermediário Crítico
Após a fragmentação, os fragmentos de ADN têm extremidades heterogéneas—alguns têm saliências 5' ou 3', alguns são retos, e alguns podem ter terminais danificados ou modificados. A etapa de reparação das extremidades utiliza uma combinação de enzimas—tipicamente a polimerase de ADN T4 (preenche saliências 5' e remove saliências 3') e a quinase de polinucleotídeos T4 (fosforila as extremidades 5')—para produzir fragmentos uniformes com extremidades retas e fosforilados a 5'.
A adição de um "A" segue-se imediatamente: uma polimerase Taq adiciona uma única adenosina à extremidade 3' de cada fragmento reto. Este "A-overhang" é complementar ao "T-overhang" único nos adaptadores padrão da Illumina, permitindo uma ligação eficiente dos adaptadores enquanto impede que os adaptadores se liguem entre si.
O parâmetro chave na reparação de extremidades é a relação entre a enzima e o DNA. Uma quantidade insuficiente de enzima deixa uma fração das extremidades não reparadas ou sem caudas de adenina, reduzindo o número de fragmentos que podem ligar-se com sucesso aos adaptadores. Isso produz uma biblioteca com um rendimento final inferior ao esperado com base na quantidade de DNA de entrada. Um sintoma comum é uma biblioteca que passa na quantificação pelo Qubit, mas apresenta baixa densidade de clusters na célula de fluxo—indicando que muitas das moléculas quantificadas carecem de sequências de adaptadores funcionais em ambas as extremidades.
A maioria dos kits de construção de bibliotecas comerciais inclui misturas de enzimas otimizadas que equilibram essas atividades. Se estiver a utilizar um protocolo personalizado, um reparo final de 30 minutos a 20°C seguido de uma adição de A de 30 minutos a 37°C é um ponto de partida padrão que pode ser ajustado com base na formulação específica da enzima.
Impacto da eficiência de A-tailing na ligadura de adaptadores a jusanteO passo de A-tailing é por vezes visto como uma reação enzimática rotineira, mas a sua eficiência determina diretamente o rendimento máximo possível de ligadura. Se apenas 80% dos fragmentos recebem uma extremidade A, a eficiência teórica máxima de ligadura cai de 50% (se cada par de fragmentos for A-tailed) para aproximadamente 40%. Esta redução de 20% em moléculas efetivas traduz-se diretamente em um menor rendimento final da biblioteca. Para amostras de baixo input onde cada molécula conta, garantir um A-tailing completo é uma das formas mais simples de maximizar o rendimento da biblioteca sem ciclos adicionais de PCR. Análise de dados genómicos a jusante também é afetado porque bibliotecas com rendimento subótimo podem produzir uma cobertura insuficiente para a deteção fiável de variantes.
Seleção de Tamanho — Um Guia Prático de Proporção de Esferas SPRI
A seleção de tamanho baseada em esferas SPRI (Imobilização Reversível em Fase Sólida) é o método mais amplamente utilizado para remover tamanhos de fragmentos indesejados de bibliotecas de NGS. O parâmetro chave é a razão entre o volume das esferas e o volume da amostra, que determina a faixa de tamanhos de fragmentos retidos. O princípio é simples: em razões de esferas mais altas, fragmentos menores são capturados; em razões de esferas mais baixas, apenas fragmentos maiores se ligam. Ao realizar duas seleções sequenciais, uma janela de tamanho alvo precisa pode ser isolada.
Compreender a relação da proporção de contasAs esferas SPRI funcionam ligando fragmentos de ADN na presença de um agente de aglomeração (PEG). A eficiência de ligação depende do tamanho: a uma dada concentração de PEG, fragmentos maiores ligam-se preferencialmente enquanto fragmentos menores permanecem em solução. Uma proporção de esferas de 0,6× significa que o volume das esferas é 60% do volume da amostra; nesta proporção, fragmentos acima de aproximadamente 500–600 bp ligam-se às esferas e podem ser capturados. O sobrenadante contém fragmentos abaixo deste limiar, incluindo a maioria dos dímeros de adaptadores. Uma proporção de 0,8× captura fragmentos acima de aproximadamente 200–300 bp. A diferença entre duas proporções consecutivas define a janela de tamanho.
| Intervalo de Fragmento Alvo | Relação da Primeira Conta (Lado Direito) | Relação do Segundo Bead (Lado Esquerdo) | Aplicação |
|---|---|---|---|
| 200–300 pb | 0,6 × (descartar contas) | 0,8× (manter contas) | Bibliotecas de amplicon e cfDNA |
| 300–500 pb | 0,6× (descartar contas) | 0,7 × (manter contas) | Bibliotecas padrão WGS, WES |
| 500–800 pb | 0,5× (descarte de contas) | 0,6 × (manter contas) | Bibliotecas de inserção longa, pares de mate. |
Como funciona a seleção de tamanho de dupla faceA primeira seleção (do lado direito) utiliza uma baixa proporção de esferas para ligar fragmentos grandes e dimers de adaptadores longos, que são descartados com as esferas. O sobrenadante contendo os fragmentos do tamanho desejado é então transferido para um segundo tubo com uma proporção de esferas mais alta para capturar os fragmentos desejados. O sobrenadante restante com fragmentos curtos e dimers de adaptadores é descartado.
A precisão da seleção de tamanho depende da diferença entre os dois rácios. Uma diferença mais ampla (por exemplo, 0,5× a 0,8×) retém uma gama de tamanhos mais ampla; uma diferença mais estreita (por exemplo, 0,6× a 0,7×) produz uma distribuição mais restrita, mas com menor rendimento total. Para a maioria das aplicações de WGS, uma seleção dupla de 0,6×/0,7× proporciona um bom equilíbrio entre rendimento e precisão. Para aplicações que requerem um controlo de tamanho mais rigoroso—como a construção de bibliotecas de cfDNA onde os fragmentos-alvo já são curtos—uma diferença mais estreita entre os dois rácios é apropriada.
Limpeza de um lado vs. limpeza de ambos os ladosUma limpeza de um só lado (relação de uma pérola, um passo de íman) apenas remove fragmentos abaixo de um limite. É mais rápida, mas não remove fragmentos grandes que podem reduzir a qualidade do cluster na célula de fluxo. Para a maioria dos protocolos de construção de bibliotecas, recomenda-se uma limpeza de dois lados para garantir que tanto os fragmentos grandes como os pequenos sejam removidos.
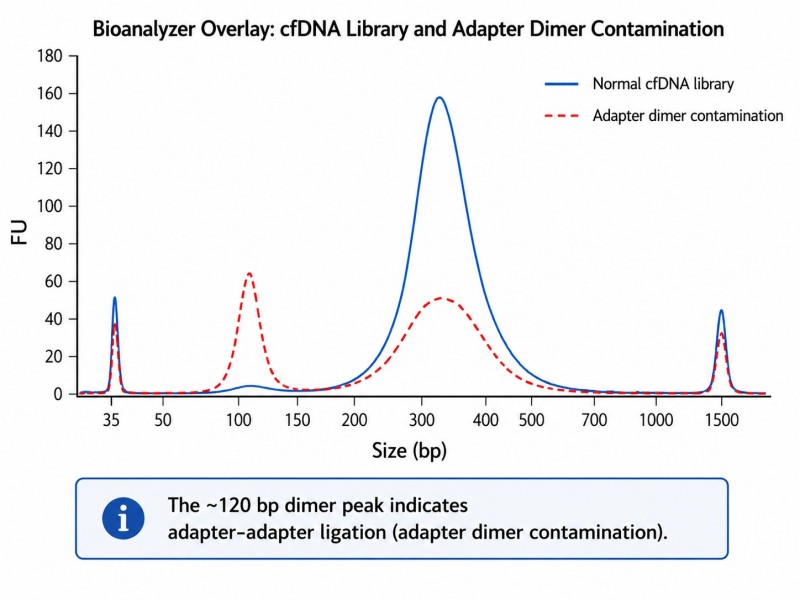 Figura 4. Guia de proporção de esferas SPRI — intervalos de seleção de tamanho de ambos os lados para diferentes tamanhos de fragmentos-alvo
Figura 4. Guia de proporção de esferas SPRI — intervalos de seleção de tamanho de ambos os lados para diferentes tamanhos de fragmentos-alvo
Legenda: Guia de seleção da proporção de esferas SPRI mostrando o protocolo de seleção de tamanhos de ambos os lados para três intervalos de fragmentos-alvo—200-300 bp, 300-500 bp e 500-800 bp—com pares de proporções de esferas correspondentes e contextos de aplicação.
Amplificação por PCR — Número de Ciclos vs. Taxa de Duplicação
A amplificação por PCR é necessária para a maioria dos fluxos de trabalho de construção de bibliotecas, mas cada ciclo adicional aumenta a taxa de duplicação e introduz viés. A contagem de ciclos ideal depende principalmente da quantidade de DNA de entrada.
A relação entre a contagem de ciclos e a duplicação não é linear. Durante os primeiros 4–6 ciclos, a amplificação está na fase exponencial, onde cada molécula de template produz uma molécula filha única, e a duplicação é mínima. Após 8–10 ciclos, a reação começa a saturar: as moléculas amplificadas começam a reanexar-se e a formar duplicados de PCR, e a taxa efetiva de geração de novas moléculas únicas diminui. Além de 10 ciclos, cada ciclo adicional adiciona aproximadamente 5–10% mais duplicados sem aumentar proporcionalmente a complexidade da biblioteca única.
Uma polimerase de alta fidelidade (taxa de erro <10⁻⁶ por base) é essencial para minimizar a introdução de mutações artefatuais durante a construção da biblioteca. A polimerase Taq padrão introduz erros a uma taxa de aproximadamente 10⁻⁴ a 10⁻⁵ por base, o que pode produzir chamadas de variantes falsas-positivas em aplicações sensíveis, como a deteção de variantes raras ou sequenciação de células únicas. As polimerases KAPA HiFi, Q5 e baseadas em Pfu são comumente utilizadas na construção de bibliotecas de NGS devido ao seu equilíbrio entre rendimento, fidelidade e viés de amplificação.
| DNA de entrada | Ciclos de PCR Recomendados | Taxa de Duplicação Esperada |
|---|---|---|
| >1 µg (para PCR livre) | 0 (sem PCR) | <5% |
| 100 ng–1 µg | 4–8 | 5–15% |
| 10–100 ng | 8–10 | 10–20% |
| 1–10 ng | 10–12 | 15–30% |
| <1 ng | 12–14 | 20–50% |
Para aplicações onde a taxa de duplicação é crítica (chamada de variantes de WGS, deteção de variantes raras), a construção de bibliotecas sem PCR é a abordagem preferida sempre que a quantidade de entrada o permita. Para amostras de baixo input onde a ausência de PCR não é viável, utilizar uma polimerase de alta fidelidade e limitar o número de ciclos ao mínimo necessário para produzir um rendimento suficiente da biblioteca é a melhor estratégia prática.
Para projetos com requisitos de cobertura específicos, serviços de sequenciação direcionada pode ajudar a otimizar os parâmetros de construção da biblioteca para a região de interesse alvo.
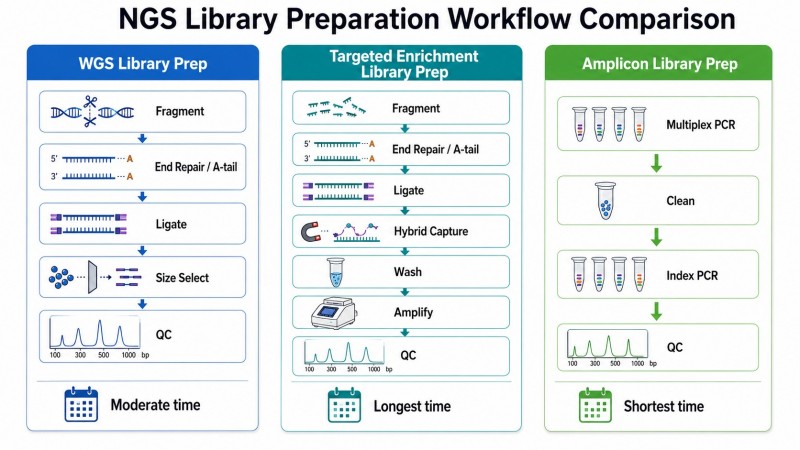 Figura 5. Contagem de ciclos de PCR vs. taxa de duplicação — relação quantitativa entre os níveis de DNA de entrada
Figura 5. Contagem de ciclos de PCR vs. taxa de duplicação — relação quantitativa entre os níveis de DNA de entrada
Relação quantitativa entre o número de ciclos de PCR e a taxa de duplicação em diferentes níveis de DNA de entrada, mostrando a transição da fase exponencial para a saturação e a faixa de contagem de ciclos ótima para cada categoria de entrada.
Juntando Tudo — Um Exemplo de Planeamento de Projecto
Para ilustrar como estes parâmetros interagem na prática, considere um investigador a planear um projeto de sequenciação do genoma humano com 48 amostras. As amostras consistem em 40 amostras de DNA sanguíneo de alta qualidade (entrada >1 µg cada) e 8 amostras tumorais FFPE (entrada ~100 ng cada, degradadas). Um único protocolo de construção de biblioteca não servirá ambos os tipos de amostras de forma otimizada.
Para as 40 amostras de DNA sanguíneo: a construção de bibliotecas sem PCR com fragmentação mecânica e uma limpeza SPRI dupla de 0,6×/0,7× é a abordagem preferida. O método sem PCR elimina o viés de duplicação e a fragmentação mecânica garante uma cobertura uniforme de GC. O tamanho alvo do inserto é de 350-500 bp, e espera-se que as bibliotecas finais produzam >5 nM por qPCR.
Para as 8 amostras FFPE: é necessária a construção de bibliotecas baseada em PCR com fragmentação enzimática (que inclui uma etapa de reparação de danos no DNA). A relação entre adaptador e inserto deve ser aumentada para 30:1 para compensar a menor entrada efetiva de DNA degradado. Os ciclos de PCR devem ser limitados a 8-10 para evitar a amplificação de artefatos. A janela de seleção de tamanho deve ser alargada (0,6×/0,8×) para acomodar a distribuição de tamanhos de fragmentos mais ampla típica das amostras FFPE.
Os dois conjuntos de bibliotecas não podem ser agrupados na mesma pista de célula de fluxo sem ajustar as concentrações de carregamento, uma vez que as bibliotecas sem PCR terão uma molaridade significativamente mais alta. A quantificação separada por qPCR para cada conjunto, seguida de um agrupamento equimolar dentro de cada conjunto, garante uma densidade de clusters consistente em todas as amostras.
Este exemplo demonstra porque os parâmetros discutidos neste guia devem ser aplicados com base em cada tipo de amostra, e não como um protocolo universal. Serviços de sequenciação NGS com a experiência na construção de bibliotecas específicas para aplicações, pode gerir estes conjuntos de parâmetros variáveis em diversos tipos de amostras dentro de um único projeto.
Construção de Bibliotecas Específicas de Aplicação — WGS vs. RNA vs. Epigenómica
Os parâmetros ótimos de construção da biblioteca diferem substancialmente dependendo da aplicação subsequente:
| Aplicação | Fragmentação | Adaptador | PCR | QC chave |
|---|---|---|---|---|
| Sequenciação do genoma completo | Mecânico ou enzimático | Adaptador Y padrão | Sem PCR ou 4–8 ciclos | Taxa de duplicação <15% |
| RNA-seq (mRNA) | Fragmentação de cDNA induzida por calor | Adaptadores específicos de RNA | 10–14 ciclos | RIN ≥ 7, eficiência de depleção de rRNA |
| Epigenómica (WGBS) | Enzimático (compatível com bisulfito) | Adaptadores metilados | 8–12 ciclos | Taxa de conversão de bisulfito >99% |
| ChIP-seq | Enzimática ou tagmentação | Adaptador Y padrão | 10–14 ciclos | Fragmentos enriquecidos ao tamanho esperado |
| Metagenómica (shotgun) | Enzimático (de baixo input) | Adaptador Y padrão | 8–12 ciclos | Sem contaminação de DNA do hospedeiro |
A principal conclusão é que não existem parâmetros de construção de bibliotecas "tamanho único". Um protocolo otimizado para WGS terá um desempenho fraco para cfDNA ou RNA-seq devido a diferenças na quantidade de entrada, distribuição do tamanho dos fragmentos e compatibilidade dos adaptadores.
Exemplo prático — Construção de biblioteca WES vs. WGSA sequenciação do exoma completo requer um passo adicional de captura por hibridização após a construção inicial da biblioteca. A biblioteca inicial deve cumprir os mesmos padrões de controlo de qualidade que uma biblioteca de WGS, mas com uma restrição adicional: a biblioteca deve ter complexidade suficiente para sobreviver ao passo de captura sem taxas excessivas de duplicação. Isso significa que a construção da biblioteca de WES deve utilizar menos ciclos de PCR do que uma biblioteca de WGS com a mesma entrada, uma vez que o passo de captura introduz uma amplificação adicional que agrava a taxa de duplicação.
Exemplo prático — construção de biblioteca de cfDNAA construção de bibliotecas de cfDNA é, sem dúvida, a mais sensível à otimização de parâmetros. A quantidade de entrada (1–50 ng) e o tamanho dos fragmentos (~167 bp) estão ambos fora da faixa ideal para protocolos padrão. A relação entre adaptador e inserto deve ser cuidadosamente controlada: se for muito baixa, a eficiência da ligação sofre; se for muito alta, a contaminação por dímeros de adaptador torna-se severa, uma vez que os curtos fragmentos de cfDNA não podem ser efetivamente separados dos dímeros por seleção de tamanho. Kits especializados de construção de bibliotecas de baixo input, com adaptadores em laço e relações de esferas otimizadas, são recomendados para fluxos de trabalho de cfDNA.
Serviços de sequenciação NGS com protocolos de construção de bibliotecas específicas para aplicações, é possível adequar o conjunto de parâmetros às necessidades de cada projeto.
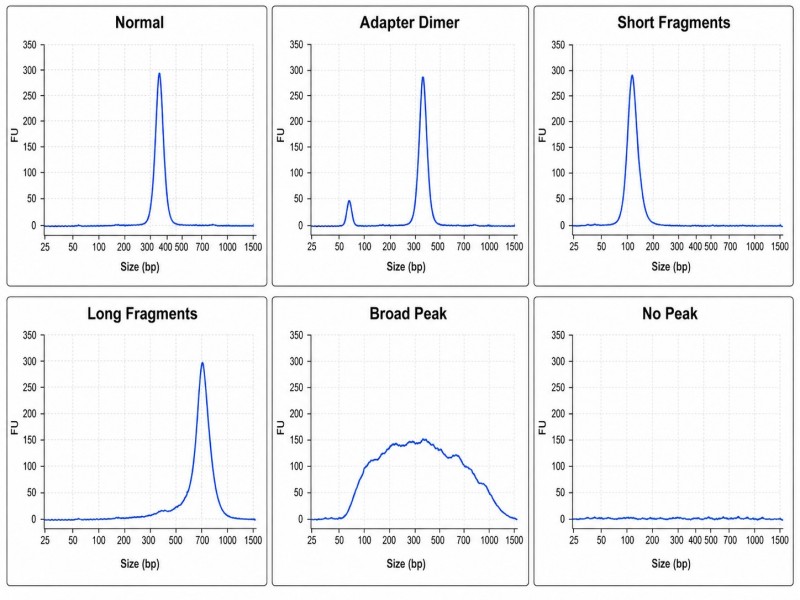 Figura 6. Construção de bibliotecas específicas de aplicação — diferenças de parâmetros em cinco aplicações comuns de NGS
Figura 6. Construção de bibliotecas específicas de aplicação — diferenças de parâmetros em cinco aplicações comuns de NGS
Tabela de parâmetros comparativos para cinco aplicações de NGS—WGS, RNA-seq, WGBS, ChIP-seq e metagenómica—mostrando diferenças no método de fragmentação, tipo de adaptador, número de ciclos de PCR e principais métricas de QC para cada aplicação.
Biblioteca QC — Critérios de Aceitação Quantificáveis
Um fluxo de trabalho de QC sistemático com limiares de aprovação/reprovação predefinidos evita que bibliotecas de baixa qualidade desperdicem capacidade de sequenciação. Os seguintes critérios representam padrões práticos para a maioria dos projetos de construção de bibliotecas Illumina. Estes limiares são baseados em experiência empírica em milhares de bibliotecas e refletem o desempenho necessário para produzir dados de sequenciação de alta qualidade em plataformas Illumina modernas.
| Métrica de QC | Método | Passar | Cuidado | Falhar |
|---|---|---|---|---|
| Concentração em biblioteconomia | qPCR | >2 nM | 1–2 nM | <1 nM |
| Conteúdo do dímero do adaptador | Bioanalisador | <5% da massa total | 5–10% | >10% |
| Tamanho médio do fragmento | Bioanalisador | Dentro de ±10% do alvo | ±10–20% | >±20% |
| Taxa de duplicação (WGS) | Computacional | <15% | 15–30% | >30% |
| qPCR vs. razão Qubit | Ambos | 0,5–2,0 | 2,0–3,0 | >3.0 |
Interpretando os resultados combinados de QCNenhum métrica única é suficiente para avaliar a qualidade de uma biblioteca. Uma biblioteca com >10% de conteúdo de dímeros de adaptadores pode ainda passar no controlo de qualidade de sequenciação se a concentração geral for alta, mas irá desperdiçar uma proporção de leituras em sequências não informativas. Uma biblioteca com baixa concentração (<1 nM) pode ainda ser carregada no chip de fluxo, mas provavelmente produzirá baixa densidade de clusters e subutilizará a capacidade do chip. O indicador mais fiável de uma boa biblioteca é aquela que passa todos os limiares de controlo de qualidade simultaneamente.
Rastreamento de lotes de enzimasUm fator frequentemente negligenciado na reprodutibilidade da construção de bibliotecas é a variabilidade entre lotes de enzimas. As enzimas utilizadas na fragmentação, reparação de extremidades, ligação e amplificação são reagentes biológicos com variabilidade inerente entre lotes. Para projetos que abrangem múltiplos lotes, é uma boa prática reter uma pequena alíquota de cada lote de enzima para comparação lado a lado. Uma amostra de controlo com desempenho conhecido deve ser incluída ao mudar para um novo lote para validar resultados consistentes.
A relação entre qPCR e Qubit é um diagnóstico particularmente útil. O qPCR mede apenas moléculas de biblioteca amplificáveis e ligadas a adaptadores, enquanto o Qubit mede todo o DNA de cadeia dupla na amostra. Se as leituras do Qubit forem significativamente mais altas do que as do qPCR (relação >3), é provável que a biblioteca contenha uma alta proporção de DNA não amplificável—tipicamente dímeros de adaptador, fragmentos não ligados ou artefatos de primers que não produzirão dados de sequenciamento. Uma relação próxima de 1 indica uma biblioteca limpa, com a maior parte do DNA medido sendo moléculas funcionais da biblioteca.
 Figura 7. Fluxograma de decisão de QC da biblioteca — critérios de aprovação/reprovação para cada ponto de controlo de QC
Figura 7. Fluxograma de decisão de QC da biblioteca — critérios de aprovação/reprovação para cada ponto de controlo de QC
Legenda: Fluxograma de decisão da QC da biblioteca mostrando os limiares de aprovação/alerta/reprovação para cinco métricas-chave—concentração de qPCR, conteúdo de dímeros de adaptadores, tamanho de fragmentos, taxa de duplicação e razão qPCR-Qubit—com ações recomendadas em cada ponto de decisão.
Como a CD Genomics Apoia a Construção de Bibliotecas
A CD Genomics oferece serviços de construção de bibliotecas de ponta a ponta, cobrindo toda a gama de métodos e tipos de amostras compatíveis com Illumina.
Métodos disponíveisO nosso laboratório executa fragmentação mecânica, fragmentação enzimática e construção de bibliotecas baseada em tagmentação, através de protocolos baseados em PCR, sem PCR e de baixo input. O método é selecionado com base na qualidade da amostra, quantidade e tipo de projeto.
Especialização em amostras especiaisValidámos protocolos de construção de bibliotecas para FFPE, cfDNA, células únicas e amostras de ultra-baixo input, com rácios de adaptadores otimizados, intervalos de seleção SPRI e contagens de ciclos de PCR para cada tipo de input.
Normas de QCCada biblioteca passa por quantificação qPCR, análise de traço no Bioanalyzer e confirmação da distribuição de tamanhos. Bibliotecas que não atendem aos critérios de aceitação descritos neste guia são sinalizadas e re-preparadas antes de prosseguir para a sequenciação.
Para mais detalhes, explore o nosso serviços de NGS ou contacte a nossa equipa para recomendações específicas do projeto.
Perguntas Frequentes
Qual é o método de fragmentação otimizado para a construção de bibliotecas de WGS?
A fragmentação mecânica (Covaris) produz a cobertura mais uniforme em relação ao conteúdo de GC com o menor viés. A fragmentação enzimática é um segundo lugar próximo e oferece maior rendimento. A tagmentação não é recomendada para WGS devido ao viés de GC em regiões de baixo GC.
Qual é a proporção de adaptador para inserto que devo usar para a construção de bibliotecas padrão?
Para entradas de 100–500 ng, recomenda-se uma razão molar de adaptador para inserto de 20:1 a 30:1. Para entradas mais altas (>500 ng), de 10:1 a 20:1; para entradas mais baixas, pode ser necessário até 50:1.
Como seleciono a proporção correta de esferas SPRI para o tamanho do meu fragmento alvo?
Para fragmentos de 300–500 bp (WGS padrão), utilize uma seleção de dois lados com uma proporção de beads de 0,6× na primeira e 0,7× na segunda. Para fragmentos de 200–300 bp (amplicon, cfDNA), utilize 0,6×/0,8×. Para fragmentos maiores (500–800 bp), utilize 0,5×/0,6×.
Quantos ciclos de PCR devo usar para a construção da minha biblioteca?
Para 100 ng–1 µg de entrada: 4–8 ciclos. Para 10–100 ng: 8–10 ciclos. Para <10 ng: 10–14 ciclos. Utilize o número mínimo que produza rendimento suficiente.
O que determina se uma biblioteca passa no controlo de qualidade?
Critérios de aprovação chave: concentração de qPCR >2 nM, conteúdo de dímeros de adaptador <5% da massa total da biblioteca, tamanho médio dos fragmentos dentro de ±10% do alvo, e razão qPCR-to-Qubit entre 0,5 e 2,0.
Como posso distinguir dímeros de adaptadores de fragmentos curtos da biblioteca em um traço do Bioanalyzer?
Os dímeros de adaptadores aparecem como um pico agudo a aproximadamente 120–140 bp. Fragmentos de biblioteca curtos produzem um pico mais amplo na mesma região, mas com uma forma diferente. Se o pico questionável for <5% da massa total, é improvável que afete significativamente o desempenho de sequenciação.
Por que é que o meu rendimento da biblioteca é inferior ao esperado?
As causas mais comuns são: concentração de DNA de entrada superestimada (use um ensaio fluorométrico, não espectrofotometria UV), relação insuficiente entre adaptador e inserto, ou limpeza excessiva das esferas que removeu material em demasia.
Posso usar o mesmo protocolo de construção de biblioteca para RNA-seq e WGS?
Não. A construção de bibliotecas de RNA-seq requer transcrição reversa, fragmentação de cDNA e incorporação de adaptadores específicos de fita. A construção de bibliotecas de WGS utiliza fragmentação de DNA seguida de ligação de adaptadores. São fluxos de trabalho fundamentalmente diferentes.
Qual é a quantidade mínima de DNA necessária para a construção de bibliotecas padrão?
Kits padrão baseados em PCR podem funcionar com entradas tão baixas quanto 0,1 ng, mas o rendimento e a complexidade tornam-se pouco confiáveis abaixo de 1 ng. Para entradas abaixo de 10 ng, recomenda-se um kit especificamente projetado para a construção de bibliotecas de baixo input.
Como é que a conversão de bisulfito afeta a construção de bibliotecas para sequenciação de bisulfito de genoma completo?
O tratamento com bisulfito converte citosinas não metiladas em uracilo, que são lidas como timinas durante o sequenciamento. Isso reduz a complexidade da sequência e requer protocolos de construção de bibliotecas especializados com adaptadores metilados e polimerases compatíveis com bisulfito.
Qual é a razão mais comum para uma baixa densidade de clusters numa biblioteca bem quantificada?
A causa oculta mais comum é a baixa eficiência de ligação dos adaptadores. A biblioteca passa na quantificação Qubit (que mede todo o dsDNA), mas muitas moléculas carecem de adaptadores funcionais em ambas as extremidades. Realizar medições de qPCR em paralelo com as medições Qubit e comparar a razão é o melhor diagnóstico.
Como escolho entre a limpeza SPRI de um só lado e a de ambos os lados para a minha biblioteca?
A limpeza de um só lado é mais rápida e suficiente para aplicações onde a remoção de dímeros de adaptadores é o objetivo principal e fragmentos grandes são menos preocupantes. A limpeza de ambos os lados é recomendada para WGS e aplicações críticas em termos de tamanho, onde tanto fragmentos grandes como pequenos devem ser controlados.
Posso usar os mesmos parâmetros de construção de biblioteca para DNA de FFPE como para DNA de tecido fresco congelado?
Não. O DNA de FFPE está tipicamente degradado (tamanho médio dos fragmentos de 200–400 bp) e contém bases danificadas. A construção da biblioteca para FFPE deve utilizar um passo de pré-reparação com uracil-DNA glicosilase, menos ciclos de PCR (para evitar a amplificação de artefatos) e uma janela de seleção de tamanho mais ampla que acomode a distribuição de fragmentos mais curtos e heterogénea.
Qual é a condição de armazenamento ideal para bibliotecas concluídas?
As bibliotecas completas devem ser armazenadas a -20°C em tubos de baixa adsorção. As bibliotecas são estáveis durante pelo menos 6 meses nessas condições. Evite ciclos repetidos de congelamento-descongelamento, aliquotando as bibliotecas em volumes de uso único.
Como posso determinar se o meu protocolo de construção de biblioteca precisa de otimização?
O indicador mais fiável é a razão qPCR-to-Qubit. Uma razão consistentemente acima de 3,0 indica que uma fração significativa do DNA quantificado não é material funcional da biblioteca. Outros sinais incluem: densidade de clusters consistentemente baixa apesar de uma concentração de carga adequada, taxas de duplicação acima de 30%, ou picos de dímeros de adaptadores acima de 10% nos traços do Bioanalyzer.
Apenas para uso em investigação.
Referências:
- Otimização de técnicas de fragmentação de DNA. Diagnósticos2025;15:2294.
- Otimização da fragmentação enzimática. BMC Genómica. 2022;23:89.
- Melhoria técnica na NGS em ambientes clínicos — otimização da ligação de adaptadores. Medicina Laboratorial Prática. 2025;40:e00430.
- Uma análise comparativa das abordagens de preparação de bibliotecas para amostras de baixo input. BMC Genómica. 2018;19:763.
- PCR multiplex de baixo número de ciclos para construção de bibliotecas. Eletroforese. 2024;45:1255-1264.


