
 Diretrizes para Submissão de Amostras
Diretrizes para Submissão de Amostras
Fluxo de Trabalho de Bioinformática de RNA-Seq
A sequenciação de RNA (RNA-seq) tornou-se uma ferramenta padrão para a análise da expressão génica, descoberta de transcritos e quantificação de splicing. O pipeline de bioinformática que processa os dados brutos de sequenciação em resultados biológicos interpretáveis é tão importante quanto a própria sequenciação—uma escolha inadequada do pipeline pode produzir chamadas de expressão diferencial enganosas, perder eventos de splicing importantes ou gerar resultados que não podem ser reproduzidos.
Este guia é escrito para investigadores que têm uma familiaridade básica com RNA-seq e precisam de orientação prática sobre como selecionar e executar o pipeline de análise adequado. Abrange as três principais abordagens de pipeline disponíveis em 2025-2026, as decisões chave em cada etapa, os requisitos computacionais a ter em conta e os erros comuns que comprometem a qualidade dos dados. O foco está no conteúdo orientado para decisões: qual pipeline escolher para um determinado tipo de projeto, quais métricas de controlo de qualidade (QC) acompanhar e como interpretar os resultados em cada fase. No final deste guia, deverá ser capaz de conceber uma estratégia completa de análise de RNA-seq que corresponda aos seus objetivos de investigação, tipos de amostras e recursos computacionais disponíveis. Para investigadores que planeiam estudos de transcriptoma em grande escala, serviços de RNA-seq incluir tanto o sequenciamento como a análise bioinformática como entregáveis integrados.
O Pipeline de Bioinformática de RNA-Seq — Três Abordagens, Um Objetivo
Cada pipeline de análise de dados de RNA-seq segue a mesma estrutura de cinco etapas: controlo de qualidade dos dados brutos, alinhamento ou quantificação de leituras, quantificação e normalização da expressão, análise estatística para expressão diferencial e interpretação funcional. As diferenças entre os pipelines residem na forma como as leituras são atribuídas a genes ou transcritos, o que determina a sensibilidade, precisão e requisitos computacionais da análise global. Compreender essas diferenças antes de iniciar a análise evita reanálises dispendiosas quando o pipeline escolhido se revela incompatível com o tipo de dados do projeto ou os objetivos de investigação.
Três abordagens de pipeline dominam a prática atual. A escolha entre elas depende da questão de pesquisa, da disponibilidade de um genoma de referência de alta qualidade e da anotação, e dos recursos computacionais disponíveis.
Caminho 1 — Alinhamento em segmentos + contagem de genes: As leituras são alinhadas a um genoma de referência utilizando um alinhador ciente de splicing (STAR ou HISAT2), e depois atribuídas a genes usando featureCounts ou HTSeq. Esta é a abordagem mais abrangente: detecta novas junções de splicing, identifica transcritos de fusão e fornece a quantificação mais precisa a nível de gene. O compromisso é o custo computacional—o alinhamento STAR de uma amostra típica de RNA-seq (30 milhões de leituras) leva de 1 a 3 horas e requer 32 GB de RAM.
Caminho 2 — Quantificação leve de transcritos: Ferramentas como Salmon, Kallisto e RSEM utilizam um transcriptoma de referência (em vez do genoma) para quantificar a abundância de transcritos diretamente a partir das leituras, contornando a etapa de alinhamento. O Salmon pode processar 30 milhões de leituras em 5-15 minutos usando 8 GB de RAM. O compromisso é que métodos leves não detectam transcritos ou junções de splicing novos e são mais adequados para projetos com um transcriptoma de referência bem anotado.
Caminho 3 — Montagem do transcriptoma: Para espécies sem um genoma de referência ou quando a descoberta de novos transcritos é o objetivo principal, ferramentas de montagem de novo (Trinity, rnaSPAdes) ou montadores guiados por genoma (StringTie, Scallop) reconstituem transcritos a partir de leituras alinhadas. Esta abordagem é a mais intensiva em termos computacionais e produz resultados que requerem validação adicional, mas é a única opção para caracterizar transcriptomas em organismos não modelo onde não está disponível um genoma de referência. O StringTie, por exemplo, pode reconstituir transcritos de comprimento completo a partir de leituras de RNA-seq alinhadas com alta sensibilidade, produzindo modelos de transcritos que podem ser utilizados para quantificação a jusante e anotação funcional.
| Tipo de Projeto | Caminho Recomendado | Métrica Chave de QC | Armazenamento por Amostra |
|---|---|---|---|
| Organismo modelo com boa anotação | STAR + featureCounts + DESeq2 | Leituras mapeadas de forma única >80% | ~15 GB (FASTQ + BAM + contagens) |
| Anotação não-modelo ou incompleta | Salmão + tximport + DESeq2 | Taxa de mapeamento >60% | ~10 GB (ficheiros FASTQ + de quantificação) |
| Descoberta de transcritos novos | STAR + StringTie + GFFcompare | Transcrições com potencial de codificação | ~25 GB (FASTQ + BAM + montagem) |
| RNA-seq de leitura longa (Iso-Seq/Nanopore) | minimap2 + TALON/Bambu | Leitura completa % | ~20-50 GB |
Para grupos de investigação que preferem externalizar a análise bioinformática, Serviços de bioinformática para RNA-seq fornecer pipelines estabelecidos para todas as três abordagens, com parâmetros personalizáveis para corresponder aos requisitos específicos do projeto.
Gestão de fluxo de trabalho para reprodutibilidade: Independentemente de qual pipeline seja escolhido, a análise deve ser executada dentro de um sistema de gestão de fluxos de trabalho, como o Nextflow (nf-core/rnaseq) ou o Snakemake. Estes frameworks garantem que os mesmos passos sejam aplicados de forma consistente em todas as amostras, que as versões do software sejam documentadas e que os resultados possam ser reproduzidos por outros investigadores. O pipeline nf-core/rnaseq, por exemplo, fornece um fluxo de trabalho de RNA-seq validado pela comunidade, com alinhamento STAR, quantificação Salmon e relatórios de QC abrangentes.
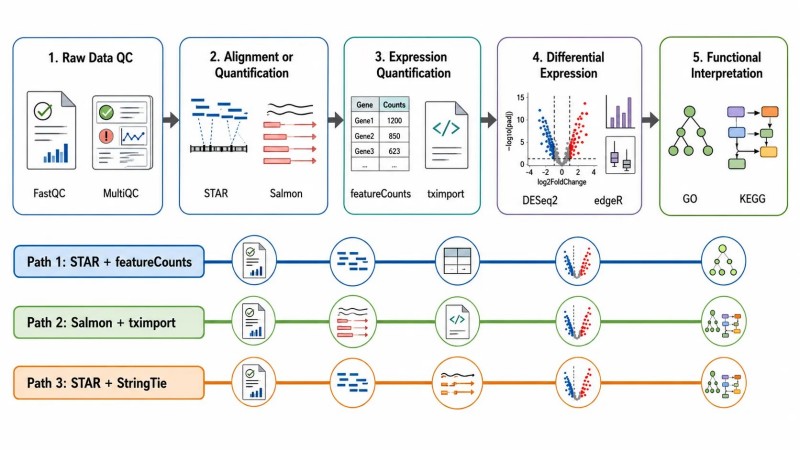 Figura 1. Pipeline de bioinformática de RNA-seq — três caminhos de análise e os seus cinco estágios partilhados
Figura 1. Pipeline de bioinformática de RNA-seq — três caminhos de análise e os seus cinco estágios partilhados
Legenda: Visão geral dos três caminhos do pipeline de bioinformática RNA-seq—alinhamento spliced + contagem de genes, quantificação leve de transcritos e montagem do transcriptoma—mostrando os cinco estágios partilhados desde a QC de dados brutos até à interpretação funcional.
Passo 1 — Controlo de Qualidade dos Dados Brutos
O controlo de qualidade é o primeiro e mais crítico passo em qualquer análise de RNA-seq. A avaliação de QC determina se os dados são adequados para a análise pretendida e identifica problemas que devem ser resolvidos antes de prosseguir.
Principais métricas de QC:
- Qualidade por base (pontuações Q): Mais de 80% das bases devem estar acima de Q30. Pontuações mais baixas indicam potenciais problemas de qualidade da corrida ou degradação da biblioteca.
- Distribuição do conteúdo GC: Deve corresponder ao conteúdo de GC esperado da espécie-alvo. A distribuição bimodal sugere contaminação ou viés da biblioteca.
- Conteúdo do adaptador: Deve ser <1% após o corte. Níveis mais altos indicam remoção incompleta do adaptador.
- Níveis de duplicação de sequência: Para RNA-seq, taxas acima de 60% sugerem baixa complexidade da biblioteca ou sobre-amplificação.
- contaminação de rRNA: Deve ser <5% para bibliotecas selecionadas por poli(A). Níveis mais altos indicam uma depleção incompleta de rRNA.
Ferramentas e fluxo de trabalho: O FastQC fornece relatórios individuais de amostras, e o MultiQC agrega-os em um único relatório para todas as amostras do projeto. O Trimmomatic e o fastp realizam o corte de adaptadores e a filtragem de qualidade — o fastp é geralmente recomendado para novos projetos, pois é mais rápido e fornece relatórios de qualidade integrados. Após o corte, uma segunda ronda de controlo de qualidade deve confirmar que o corte foi eficaz sem cortar em excesso sequências biologicamente significativas. Se o controlo de qualidade revelar problemas significativos em uma ou mais amostras (por exemplo, contaminação de rRNA >20%, mediana do Q-score <30), as amostras afetadas devem ser sinalizadas antes de prosseguir para o alinhamento.
Estratégia de QC por amostra vs. estratégia de QC multi-amostra: Uma recomendação comum é executar o FastQC em cada amostra individualmente e, em seguida, usar o MultiQC para agregar os resultados de todo o projeto. Esta abordagem revela tanto problemas específicos de amostra como problemas sistemáticos. O mapa de calor por amostra no MultiQC é particularmente útil para identificar amostras atípicas que devem ser examinadas mais de perto antes da inclusão na análise DE.
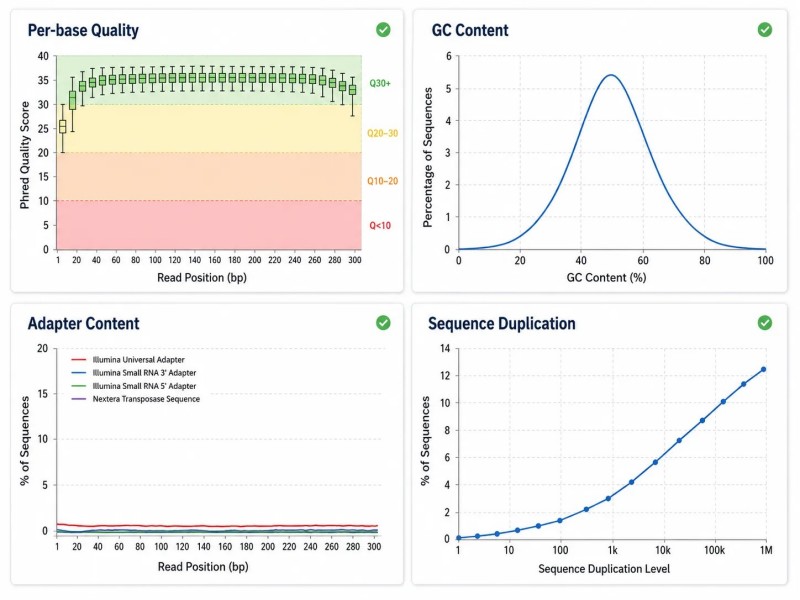 Figura 2. Relatório FastQC — métricas chave para a avaliação da qualidade de dados de RNA-seq
Figura 2. Relatório FastQC — métricas chave para a avaliação da qualidade de dados de RNA-seq
Relatório de qualidade FastQC para dados de RNA-seq mostrando pontuações de qualidade por base, distribuição de conteúdo de GC, conteúdo de adaptadores, níveis de duplicação de sequências e métricas de contaminação de rRNA com limites recomendados.
Passo 2 — Escolher entre Alinhamento, Quantificação Leve e Montagem
A decisão central em um pipeline de RNA-seq é como atribuir leituras a transcrições ou genes. Existem três abordagens fundamentalmente diferentes, cada uma com compromissos distintos em termos de precisão, custo computacional e capacidade de descoberta.
STAR (alinhamento em segmentos): o STAR utiliza um algoritmo de busca sequencial de sementes mapeáveis máximas que é significativamente mais rápido do que alinhadores anteriores como o TopHat ou o Bowtie2. Ele detecta junções de splicing canónicas e não canónicas, leituras quiméricas para deteção de fusões, e fornece alinhamentos que podem ser utilizados tanto para quantificação como para montagem de transcritos. Para projetos com requisitos específicos de alinhamento, serviços de NGS pode ser configurado para fornecer dados em formatos compatíveis com STAR, Salmon ou outros pipelines de análise.
HISAT2 (indexação hierárquica): O HISAT2 é um alinhador alternativo que reconhece splicing, utilizando um índice hierárquico baseado em FM-index, requerendo menos memória (4-8 GB por amostra) do que o STAR. É viável quando ambientes com restrições de memória impedem o uso do STAR, embora detecte, em média, menos junções de splicing novas.
Salmão (quantificação leve): O Salmon utiliza uma abordagem de quasi-mapeamento que determina a origem do transcrito mais provável para cada leitura sem realizar um alinhamento completo. Corrige o viés de GC e o viés específico de sequência, produzindo estimativas de TPM a nível de transcrito em minutos.
StringTie (montagem de transcritos): O StringTie monta transcritos a partir de leituras alinhadas, produzindo um modelo de transcrito para cada lócus. Pode ser utilizado com ou sem uma anotação de referência. Para projetos que requerem descoberta de transcritos de novo, o StringTie seguido pela fusão entre amostras produz um transcriptoma abrangente que captura isoformas novas.
Estrutura de decisão: Para projetos de referência de alta qualidade, o STAR + featureCounts fornece a quantificação a nível de gene mais abrangente. Para projetos grandes que priorizam a eficiência, o Salmon oferece uma precisão comparável a uma fração do tempo de computação. Para descoberta de transcritos ou espécies não-modelo, a montagem guiada por genoma com o StringTie é apropriada. Para deteção de fusões, Serviços de análise de RNA-seq pode incorporar o STAR-Fusion ou o Arriba.
Consideração prática — execução de múltiplos pipelines: Para projetos de alta prioridade, executar tanto o STAR como o Salmon e comparar os resultados fornece validação interna. Se ambas as pipelines classificarem os mesmos genes como os principais candidatos, a confiança é substancialmente maior do que a proveniente de uma única pipeline.
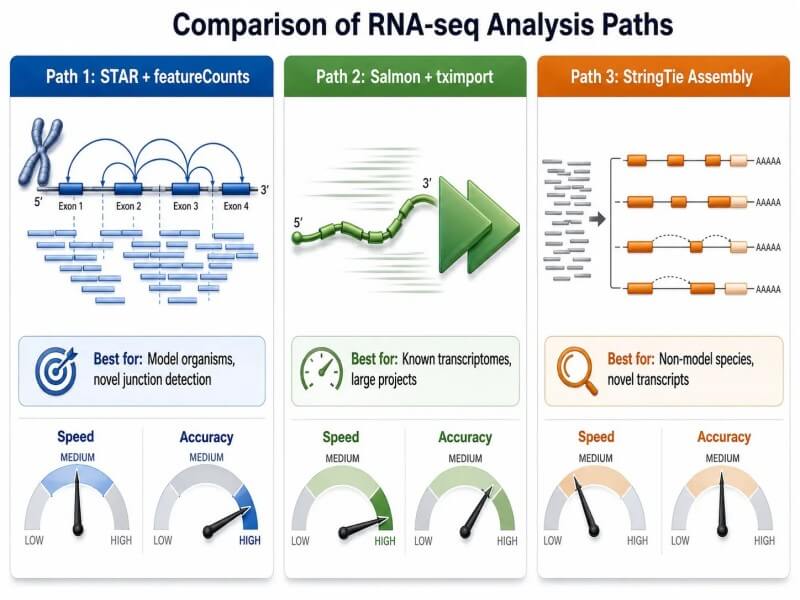 Figura 3. Três caminhos de análise de RNA-seq — alinhamento, quantificação leve e montagem de transcritos
Figura 3. Três caminhos de análise de RNA-seq — alinhamento, quantificação leve e montagem de transcritos
Diagrama comparativo de três caminhos de análise de RNA-seq mostrando o fluxo de dados através do alinhamento STAR + featureCounts, quantificação leve do Salmon e montagem de transcritos do StringTie, com principais compensações em precisão, custo computacional e capacidade de descoberta.
Passo 3 — Quantificação e Normalização da Expressão
A quantificação converte leituras alinhadas ou fragmentos mapeados em medidas de expressão ao nível do gene ou transcrito. A escolha do método de normalização tem um impacto direto nos resultados de expressão diferencial e na comparabilidade entre amostras.
Quantificação a nível de gene com featureCounts: O featureCounts conta as leituras que sobrepõem os exões de cada gene, produzindo uma matriz de contagem bruta. Esta é a entrada padrão para o DESeq2 e o edgeR. O parâmetro chave é a configuração de especificidade de fita: para bibliotecas com fita, especificar a correta orientação da fita duplica o sinal utilizável.
HTSeq-count: Uma alternativa ao featureCounts utilizando modelos de interseção baseados em Python. É mais lento, mas oferece mais controlo sobre a lógica de atribuição de leituras para modelos genéticos complexos com anotações sobrepostas.
Quantificação a nível de transcrito com Salmon: O Salmon produz contagens estimadas e valores de TPM por transcrito. Para a análise a nível de gene, o tximport agrega as estimativas de transcritos a nível de gene, tendo em conta as diferenças de comprimento dos transcritos. A correção de viés incorporada no Salmon reduz erros sistemáticos.
Métodos de normalização: TPM normaliza tanto a profundidade de sequenciação como o comprimento do transcrito, sendo apropriado para comparações dentro da amostra. A mediana das razões do DESeq2 normaliza apenas para a profundidade e é o padrão para a análise de DE entre amostras. FPKM e RPKM são métodos mais antigos, em grande parte substituídos pela normalização TPM e DESeq2.
Qual normalização usar quando: Para comparações dentro da amostra, o TPM é apropriado. Para análises de DE entre amostras, deve-se usar DESeq2 ou normalização TMM. Para meta-análises que combinam múltiplos estudos, o TPM é preferido, pois é menos sensível a diferenças de tamanho de biblioteca.
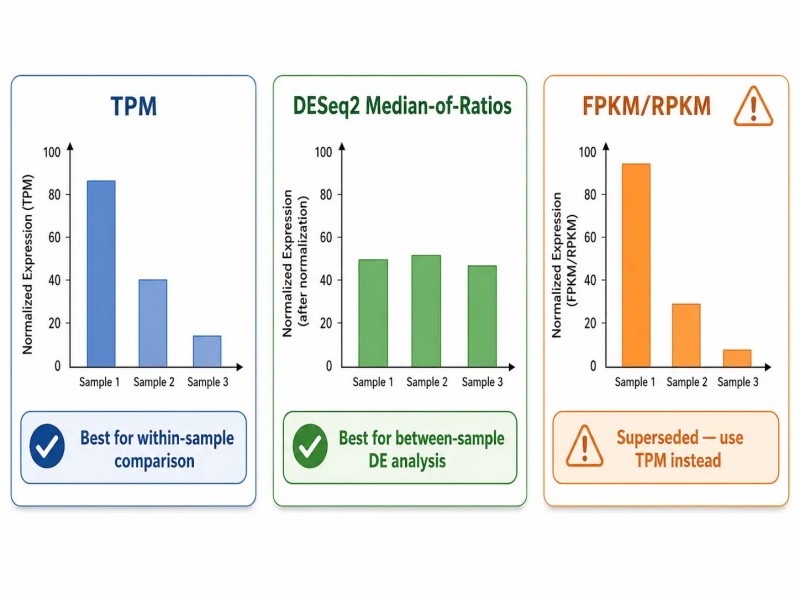 Figura 4. Métodos de normalização — TPM, mediana de razões DESeq2 e FPKM comparados.
Figura 4. Métodos de normalização — TPM, mediana de razões DESeq2 e FPKM comparados.
Legenda: Comparação de TPM, median-of-ratios do DESeq2 e normalização FPKM mostrando fórmulas, casos de uso (dentro da amostra vs entre amostras) e vieses resultantes de uma escolha incorreta de normalização.
Passo 4 — Análise de Expressão Diferencial
A análise de expressão diferencial (DE) identifica genes cuja expressão muda significativamente entre condições. A escolha do método estatístico afeta o poder de detectar verdadeiros positivos, o controlo de falsos positivos e a capacidade de lidar com as características dos dados de RNA-seq.
Três métodos convencionais:
- DESeq2: Modelo binomial negativo com estimador de dispersão por encolhimento. Desempenha bem com tamanhos de amostra pequenos (3-4 réplicas) e proporciona um controlo robusto da FDR. A deteção de outliers de contagem incorporada reduz os falsos positivos.
- edgeR: Modelo binomial negativo com estimativa de dispersão empírica de Bayes. Ligeiramente mais sensível para conjuntos de dados com replicados maiores (>6 por grupo). O quadro de quasi-verossimilhança proporciona um controlo de FDR mais conservador.
- limma-voom: Estrutura de modelagem linear com pesos de precisão. Computacionalmente eficiente, resultados comparáveis ao DESeq2 para a maioria dos conjuntos de dados. Bem adequada para desenhos experimentais complexos com múltiplos fatores.
Com apenas duas réplicas, a análise DE pode ser realizada, mas o controlo do FDR é pouco fiável. A saída padrão é uma lista de genes classificados por valor p ajustado e log2 de mudança de fold, tipicamente filtrada a FDR < 0,05 e |log2FC| > 1. Para a análise padrão, serviços de análise de dados genómicos fornecer pipelines validados DESeq2 e edgeR.
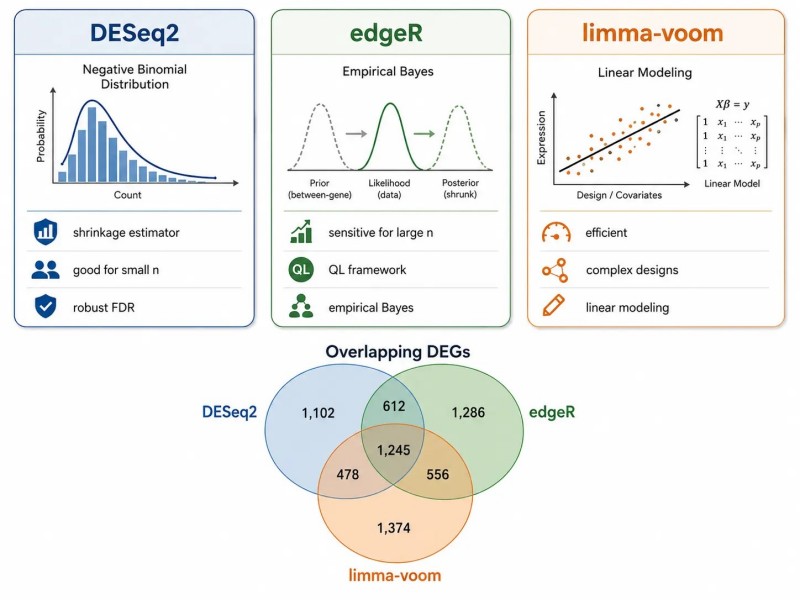 Figura 5. Métodos de expressão diferencial — DESeq2, edgeR e limma-voom comparados
Figura 5. Métodos de expressão diferencial — DESeq2, edgeR e limma-voom comparados
Legenda: Comparação entre DESeq2, edgeR e limma-voom mostrando modelos estatísticos, estimativa de dispersão, tamanhos de amostra ótimos e melhores casos de uso para cada método.
Interpretação Funcional — De Listas de Genes a Mecanismos Biológicos
Uma vez identificados os genes diferencialmente expressos (DEGs), três abordagens complementares interpretam o seu significado biológico:
- Análise de sobre-representação (ORA): Testa o enriquecimento de DEG em categorias GO ou vias KEGG em relação ao fundo. Requer um limiar de significância e é sensível ao corte escolhido.
- Análise de enriquecimento de conjuntos de genes (GSEA): Utiliza uma lista de genes totalmente ordenada para testar a enriquecimento no topo ou na parte inferior da classificação. Não requer um limiar e deteta alterações coordenadas mesmo quando genes individuais não são significativos.
- Análise de vias e redes: Ferramentas como IPA, Metascape e clusterProfiler mapeiam os DEGs em vias biológicas. A saída inclui previsões de ativação de vias e de reguladores upstream.
O controlo de versão da base de dados é essencial para a reprodutibilidade — as bases de dados GO, KEGG e Reactome são atualizadas regularmente. Um erro comum é usar uma versão da base de dados incompatível com a versão da anotação utilizada durante o alinhamento. Serviços de bioinformática personalizados pode incorporar a análise de enriquecimento de vias e análise de redes para projetos que exigem uma profundidade adicional.
Bioinformática de RNA-Seq de Célula Única — Um Paradigma Diferente
A bioinformática de RNA-seq de célula única (scRNA-seq) difere fundamentalmente da análise em massa. Os dados são muito mais escassos e o ruído técnico proveniente de eventos de dropout é o principal desafio. O fluxo de trabalho padrão de scRNA-seq inclui: processamento de dados brutos (CellRanger), filtragem de qualidade, normalização (SCTransform, scran), redução de dimensionalidade (PCA, UMAP), agrupamento (Louvain, Leiden), identificação de genes marcadores e anotação de tipos celulares.
Os requisitos computacionais são substancialmente mais elevados: um conjunto de dados de 10.000 células requer 16-32 GB de RAM. Um conjunto de dados de 100.000 células pode exigir 64 GB ou mais. Recomenda-se o uso de nuvem ou clusters de HPC para conjuntos de dados que excedam 50.000 células. Serviços de bioinformática fornecer pipelines padronizados para análise de bulk e scRNA-seq.
Requisitos Computacionais para Análise de Dados de RNA-Seq
O planeamento de recursos computacionais antes da chegada dos dados de sequenciação previne gargalos na análise. Para um projeto de RNA-seq em bulk com 48 amostras (30M leituras cada, 2x150 bp):
- Armazenamento: ~430 GB de FASTQ bruto + 250-400 GB de BAM + 10-50 GB de ficheiros intermédios = 700-900 GB no total. FASTQ comprimido reduz os requisitos em 40-60%.
- Tempo de computação: Alinhamento STAR 48-144 hr de núcleo único (paralelizado). Quantificação Salmon 4-12 hr. DESeq2 30 min-2 hr. Montagem StringTie 24-72 hr.
- Memória: STAR 32 GB por amostra. DESeq2/edgeR 8-16 GB. Salmon 4-8 GB. scRNA-seq (10K células) 16-32 GB.
- Infraestrutura: 64+ GB de RAM, 16+ núcleos de CPU, armazenamento SSD recomendado. A análise na nuvem é mais rentável para projetos com mais de 100 amostras. Para laboratórios sem HPC, Fornecedores de serviços de WGS e RNA-seq tipicamente incluem análise computacional.
Consideração de estrandamento: A maioria dos projetos modernos de RNA-seq utiliza preparação de bibliotecas com orientação. As configurações do pipeline (por exemplo, a flag de orientação do featureCounts) devem corresponder ao tipo de biblioteca para evitar a perda de metade do sinal devido a leituras atribuídas incorretamente.
 Figura 6. Erros comuns em RNA-seq — problemas, causas e soluções
Figura 6. Erros comuns em RNA-seq — problemas, causas e soluções
Legenda: Erros comuns na análise de RNA-seq, incluindo DEGs excessivos, efeitos de lote, baixas taxas de mapeamento, réplicas insuficientes e irreproduzibilidade do pipeline.
Erros Comuns na Análise de RNA-Seq e Como Evitá-los
| Problema Observado | Causa Raiz | Prevenção |
|---|---|---|
| DEGs excessivos (>30% dos genes) | Normalização inadequada ou limiar de FDR | Plano de análise de pré-registo; verificar estimativas de dispersão |
| Os efeitos de lote dominam a variação. | Amostras processadas em ordem não aleatória | Aleatorizar o processamento; incluir lote na matriz de design. |
| Taxa de mapeamento baixa (<60%) | Desajuste de referência, contaminação de rRNA, degradação | Verificar referência; verificar o conteúdo de rRNA no QC. |
| Sem DEGs significativos apesar das diferenças visíveis. | Replicados insuficientes, alta variabilidade dentro do grupo | Mínimo de 3 réplicas; considerar aumentar o tamanho da amostra. |
| Resultados irreproduzíveis entre pipelines | Diferentes normalizações ou pressupostos estatísticos | Utilize pelo menos dois métodos e compare a concordância. |
Perguntas Frequentes
Qual é a melhor ferramenta de alinhamento para RNA-seq?
O STAR é o alinhador ciente de splices mais amplamente utilizado para RNA-seq. É significativamente mais rápido que o TopHat2 e o HISAT2 para a maioria dos conjuntos de dados e detecta mais junções de splice novas.
Devo usar o STAR ou o Salmon para a quantificação de RNA-seq?
Escolha o STAR para novos junctions de splicing, deteção de fusões ou montagem de transcritos. Escolha o Salmon para quantificação rápida de transcritos conhecidos em grandes projetos. Muitos projetos beneficiam-se do uso de ambos.
Quantos replicados biológicos preciso para RNA-seq?
Mínimo de três réplicas biológicas por condição para uma análise de DE fiável. Com menos, o controlo do FDR não é fiável. Para uma alta variabilidade biológica, podem ser necessárias 4-6 réplicas.
Qual é a diferença entre FPKM, RPKM e TPM?
Todos normalizam para a profundidade e comprimento do transcrito, mas o TPM é preferido. O FPKM e o RPKM introduzem viés nas comparações entre amostras. O TPM normaliza o comprimento antes do tamanho da biblioteca para uma comparabilidade direta entre amostras.
Como posso lidar com efeitos de lote em dados de RNA-seq?
Abordagem na fase de design, randomizando amostras entre lotes. Incluir o lote como covariável na matriz de design do DESeq2 ou limma. A correção computacional (RUVseq, sva, ComBat-seq) pode reduzir os efeitos, mas não consegue resolver a confusão completa.
Apenas para fins de investigação, não se destina a diagnóstico clínico, tratamento ou avaliações de saúde individuais.
Referências:
- Do banco aos bytes: um guia prático para a análise de dados de RNA-seq. Fronteiras em Genética. 2025;16:1697922.
- Um fluxo de trabalho abrangente para otimizar a análise de dados de RNA-seq. BMC Genómica. 2024;25:678.
- Avaliação de desempenho das plataformas de sequenciação de DNA no estudo da ABRF. Biotecnologia da Natureza. 2021;39:1348-1365.
- Análise diferencial de transcritos mais rápida e precisa com edgeR v4 e Salmon. Genómica e Bioinformática NAR. 2024;6(4):lqae151.
- Análise de expressão diferencial DESeq2. Bioconductor (versão atual).


